Table of contents
1. Introduction
Here, we introduce the Differential Expression Workflow Executor (DEWE) for RNA-Seq software, or simply DEWE, a bioinformatic tool to perform genetic differential expression analysis of RNA-Seq data.
DEWE incorporates pre-cooked workflows for RNA-Seq differential expression analysis which are able to compare two conditions with at least two samples each. Furthermore, DEWE also offers a simple, user-friendly GUI for common individual operations carried out in RNA-Seq data analysis such as quality control, reads alignment, and so on.
Additionally, a full PDF manual is available here.
1.1 Third party software
Table 1 shows the specific third-party tools and versions that DEWE uses for each step of the analysis.
Table 1: Third-party software used in DEWE.
Table of contents
- 2. Installation
2. Installation
DEWE is available under two different installation methods: through a Docker container or through the use of a Virtual Box Machine.
The main advantages of using Docker is that the user can forget to install all the necessary dependencies to run DEWE, as well as having to execute updating tasks of both DEWE and its dependencies, this will be done completely automatically.
As it runs within a controlled environment such as Docker, its installation is much simpler for the final user.
In addition, a VirtualBox Virtual Machine with DEWE and all its dependencies is available for download.
2.1 Docker installers
The Docker installers are available for the following operating systems:
- Windows 7 or higher
- Linux with 3.10 kernel minimum
- Mac OS X "Mountain Lion" or newer
2.1.1 Windows Installer
WARNING: this is a beta version, since it is a non-stable version, errors may occur during execution.
2.1.1.1 Prerequisites
To install DEWE, your machine must have a 64-bit operating system running Windows 7 or higher. Additionally, you must make sure that virtualization is enabled on your machine.
If virtualization is not enabled on your system, follow the manufacturer’s instructions for enabling it.
Depending on your system configuration you may need to pause the antivirus software to install correctly this software.
The DEWE installer for Windows is available at the following link http://static.sing-group.org/software/DEWE/installers/1.2/DEWE-windows-1.2.exe.
2.1.1.2 Installation
After executing the installer, you have to select the shared folder with DEWE. This folder will be accessible from DEWE. Other folders outside this one will not be available.


Depending on the configuration of the Operating System, it may be necessary to accept some alerts and the installation of additional drivers.
Once the setup has completed, the computer will not only have DEWE installed, but a fully functional VirtualBox, Docker and Xpra client applications.

2.1.1.3 First run
DEWE is now ready to start working. You can open it from the DEWE icon on the desktop and also from the Start Menu folder.
The first time it will take a while to start because it has to download the last version available of the DEWE docker image (~600MB) and execute it.
2.1.1.4 DEWE Viewer
As we have seen in the previous section, to execute DEWE in Windows the user has to execute the DEWE shortcut.
As the start of the tool consumes certain amount of time, at the time of closing DEWE the user has two options. On the one hand, let it run in the background, and on the other hand close it completely. If it has been decided to let it run in the background, the user can reduce considerably the time to restart the application using the DEWE Viewer shortcut. This shortcut will do, instead of rebooting DEWE, move the tool from second to foreground. If, on the contrary, the tool has been completely closed, the DEWE shortcut must be used to restart the tool.
2.1.1.5 Uninstallation
DEWE can be uninstalled using Add/Remove Programs from Control Panel. You can uninstall DEWE and the main dependencies separately: VirtualBox, Xpra and Docker Toolbox.
2.1.2 Linux Installer
2.1.2.1 Prerequisites
DEWE requires a 64-bit installation regardless of the computer Linux version. Additionally, the computer kernel must be 3.10 at minimum. To check the current kernel version, open a terminal and use uname -r to display your kernel version.
The DEWE installer for Linux is available at the following link http://static.sing-group.org/software/DEWE/installers/1.2/DEWE-linux-1.2.sh.
Also, the DEWE uninstaller is available at http://static.sing-group.org/software/DEWE/installers/1.2/Uninstall-DEWE-linux-1.2.sh.
2.1.2.2 Installation
On the installer download folder open a terminal and execute this command as root:
$ sh ./install-dewe.sh
To be able to share files with DEWE, a path must be selected.
The installer will ask for a path, by default /home will be selected as shared directory.

Once the setup has completed, the computer will not only have DEWE installed, but a fully functional Docker and Xpra client applications.
2.1.2.3 First run
DEWE is now ready to start working. You can open it from the DEWE icon on the Start Menu or typing on terminal:
$ dewe
The first time it will take a while to start because it has to download the last version available of the DEWE docker image (~600MB) and execute it.
2.1.2.4 Uninstallation
On the uninstaller download folder open a terminal and execute this command as root:
$ sh ./uninstall-dewe.sh
The uninstaller will prompt you to uninstall the dependencies of Docker and XPRA before starting the DEWE uninstallation process.
Once the uninstaller execution is complete, DEWE (and its dependencies if selected) have been removed from the computer.
2.1.3 OS X Installer
WARNING: this is a beta version, since it is a non-stable version, errors may occur during execution.
2.1.3.1 Prerequisites
To install DEWE, your Mac must be running OS X 10.8 "Mountain Lion" or newer.
The DEWE installer for Mac OS is available at the following link http://static.sing-group.org/software/DEWE/installers/1.2/DEWE-MacOSX-1.2.pkg.zip.
2.1.3.2 Installation
Execute the installer and accept the terms of the installation. Once the setup has completed, you will not only have DEWE installed, but a fully functional VirtualBox, Docker and Xpra client applications.

2.1.3.4 First run
By opening the DEWE application from the Applications folder, Terminal window will be opened where you can see the progress of the configuration and the download of the Docker image (~600MB). The download of the Docker image is only required the first time you run DEWE and may take a while, depending on your network connection.

2.1.3.3 Uninstallation
You can uninstall DEWE and the main dependencies separately: VirtualBox, Xpra and Docker Toolbox. To uninstall them just drag the app from the Applications folder to the Trash.
2.1.4 Docker installers FAQ
This section contains the Frequently Asked Questions about DEWE and several known problems that may occur when using the application:
2.1.4.1 Windows installer
2.1.4.1.1 Error checking TLS connection
Error checking TLS connection: Error getting driver URL: Something went wrong running an SSH command!
This error may happen the first time DEWE is started and is related to the network configuration of VirtualBox. To fix it, it is necessary to check the network configuration in VirtualBox.
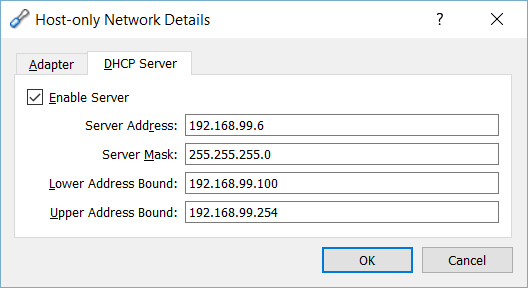
Open VirtualBox and go to menu File > Preferences. Then select Network section and go to Host-only Networks tab. Select "VirtualBox Host-Only Ethernet Adapter #2" and edit its configuration. On the DHCP Server tab, DHCP service must be enabled with the following configuration:
- Server Address: 192.168.99.6
- Server Mask: 255.255.255.0
- Lower Address Bound: 192.168.99.100
- Upper Address Bound: 192.168.99.254
2.1.4.1.2 Errors ocurred. See the logfile xpra.exe.log for details
Errors ocurred. See the logfile xpra.exe.log for details
This error may occur because there are some permission problems in the installation path of Xpra on Windows 10.
The current workaround is to use the shortcut DEWE Viewer available in the Start Menu.
2.1.4.1.3 Errors occurred
Errors occurred: See the logfile 'C:\Program Files (x86)\Xpra\xpra.exe.log' for details
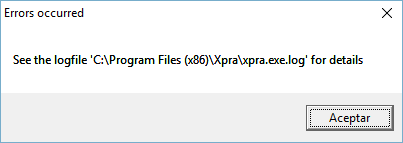
This error may happen every time DEWE is started in Windows and it is related to a permissions problem when starting the Xpra client.
The current workaround is to use the DEWE Viewer shortcut available in the folder DEWE under All programs.
2.1.4.2 MAC OS X installer
2.1.4.2.1 Installer can't be opened because it is from an unidentified developer
dewe-installer.pkg can't be opened because it is from an unidentified developer. Your security preferences allow installation of only apps from the Mac App Store and identified developers.
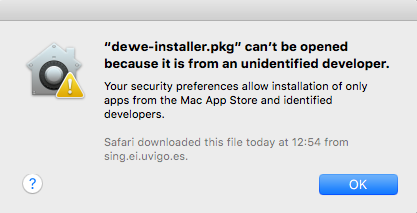
In this case, you need to allow the execution of the installer in the system settings: "Security & Privacy". There, click the button "Open Anyway" to launch the installer, or allow apps downloaded from Anywhere.

2.1.4.2.2 Some characters are missing in Mac version
In the Mac OSX version of DEWE there are some incompatibilities with several keyboard layouts like ES-ISO. Some keys may print spaces instead of the character associated to the key. The current workaround is to use another keyboard layout like ES or EN-US.
2.1.4.3 Linux installer
2.1.4.3.1 Client is newer than server
docker: Error response from daemon: client is newer than server (client API version: 1.22, server API version: 1.21). See 'docker run --help'.
This error may occur when updating DEWE. In this case, you need to upgrade your docker image using the following command:
$ docker-machine upgrade
Once it finishes the upgrade, you need to restart DEWE virtual machine from VirtualBox or reboot your system.
2.2 Virtual machine
2.2.1 Prerequisites
VirtualBox is needed to use the DEWE Virtual Machine. The installation files and instructions of this programs can be found in VirtualBox.org.
2.2.2 Download
The DEWE Virtual Machine is available at the following link http://static.sing-group.org/software/DEWE/installers/1.2/DEWE-VM-1.2.vdi.
2.2.3 Installation
Decompress the downloaded file (you will need a ZIP decompressor).
Once decompressed you should have a file called DEWE-VM.vdi with is the virtual machine hard disk.
Now you are ready to use the DEWE Virtual Machine.







2.2.4 First run
DEWE is now ready to start working. You can open it from the DEWE icon on the Start Menu:

$ dewe
The virtual machine password is: dewepass
Table of contents
- 3. DEWE workflows
- 3.1 Quality control [manual]
- 3.2 Bowtie2, StringTie and R packages (Ballgown and edgeR)
- Step 1: download the dataset
- Step 2: configure the workflow
- Step 3: import the reference genome index
- Step 4: reference genome selection
- Step 5: introducing the experimental conditions
- Step 6 : samples selection
- Step 7: reference annotation file selection
- Step 8: working directory selection
- Step 9: parameter configuration
- Step 10: workflow configuration summary
- Step 11: monitoring the workflow execution
- Step 12: workflow results
- 3.3 HISAT2, StringTie and R packages (Ballgown and edgeR)
- Step 1: download the dataset
- Step 2: configure the workflow
- Step 3: import the reference genome index
- Step 4: reference genome selection
- Step 5: introducing the experimental conditions
- Step 6 : samples selection
- Step 7: reference annotation file selection
- Step 8: working directory selection
- Step 9: parameter configuration
- Step 10: workflow configuration summary
- Step 11: monitoring the workflow execution
- Step 12: workflow results
- 3.4 Configure a workflow using the workflow.dewe file
- 3.5 Workflow execution FAQ
3. DEWE workflows
Currently, DEWE provides two differential expression analysis workflows:
- HISAT2, StringTie and R packages (Ballgown and edgeR)
- Bowtie2, StringTie and R packages (Ballgown and edgeR)

DEWE also includes two DE analysis libraries, Ballgown and edgeR, offering the end-user the possibility to compare gene/transcript expression results through two different normalisation procedures (Fragments Per Kilobase Million or FPKM vs Trimmed Mean of M-values or TMM). Although most of the DEWE outputs, mainly tables and figures, are originated from Ballgown, edgeR remains a tool widely used in the scientific community. For this reason, we consider interesting keeping this analysis and bringing the user the possibility to compare their results with many other pipelines that still use edgeR.
It is important to note that quality control operations must be performed manually by the user before the workflow execution, thus they are not included in the built-in workflows.
Additionaly, and for a detailed review of the basic conceps of RNA-Seq analysis, the reader is prompted to look up the Galaxy Project tutorial: http://galaxyproject.github.io/training-material/topics/transcriptomics/tutorials/rb-rnaseq/tutorial.html.
The available workflows are accessible from the Workflow catalog. This section contains tutorials for configure and execute the available workflows using real datasets.
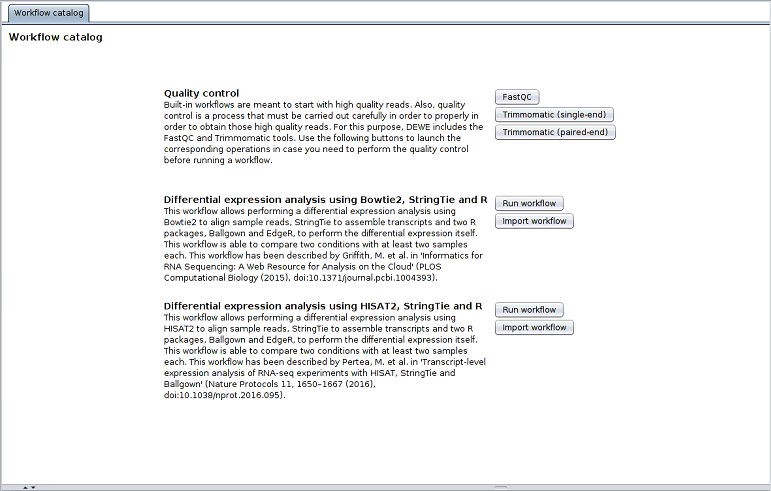
3.1 Quality control [manual]
Before executing a workflow, users may need to perform quality control analysis and reads filtering in order to feed the workflows with proper data. For this task, DEWE offers the FastQC tool for the quality control of the samples, and Trimmomatic to perform reads filtering. It is important to note that these quality control operations must be performed manually by the user before the workflow execution, thus they are not included in the built-in workflows.
3.1.1 FastQC
DEWE allows the generation of a FastQC quality control report for multiple reads files. Clicking on the FastQC button in the Workflow catalog, a new window will be displayed and the following data will be requested:
- Input files: the reference annotation file (.gtf).
- Output directory: optionally, the directory where the reports must be generated. If not provided, the output report for each reads file is created in the same directory as the reads file being processed.

3.1.2 Timmomatic
DEWE provides operations for performing reads filtering using Trimmomatic.
3.1.2.1 Single-end reads filtering
This operation allows filtering single-end raw reads using Trimmomatic. Clicking on the Trimmomatic (single-end) button in the Workflow catalog, a new window will be displayed and the following data will be requested:
- Input file: the input reads file.
- Trimmomatic parameters: the steps for trimmomatic and its configuration. The Steps tab allows selecting which steps must be applied and define the order in which they should be applied. Then, the other tabs allows configuring each step. The following steps are available:
- Leading: removes low quality bases from the beginning. As long as a base has a value below this threshold the base is removed and the next base will be investigated.
- Trailing: removes low quality bases from the end. As long as a base has a value below this threshold the base is removed and the next base (which as trimmomatic is starting from the 3' prime end would be base preceding the just removed base) will be investigated. This approach can be used removing the special illumina 'low quality segment' regions (which are marked with quality score of 2), but we recommend Sliding Window or MaxInfo instead.
- Crop: removes bases regardless of quality from the end of the read, so that the read has maximally the specified length after this step has been performed. Steps performed after CROP might of course further shorten the read.
- Headcrop: removes the specified number of bases, regardless of quality, from the beginning of the read.
- Minlen: removes reads that fall below the specified minimal length. If required, it should normally be after all other processing steps. Reads removed by this step will be counted and included in the „dropped reads‟ count presented in the trimmomatic summary.
- Sliding window: performs a sliding window trimming, cutting once the average quality within the window falls below a threshold. By considering multiple bases, a single poor quality base will not cause the removal of high quality data later in the read.
- Max info: performs an adaptive quality trim, balancing the benefits of retaining longer reads against the costs of retaining bases with errors.
- Quality: reencodes the quality part of the FASTQ file to the selected base.
- Illumina clip: finds and removes Illumina adapters.
- Output directory: optionally, the directory where the filtered file must be created. If not provided, the output file is created in the same directory as the reads file being filtered.
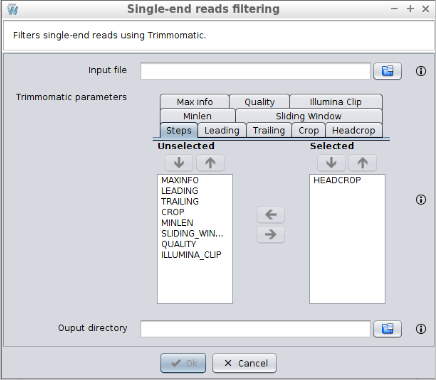
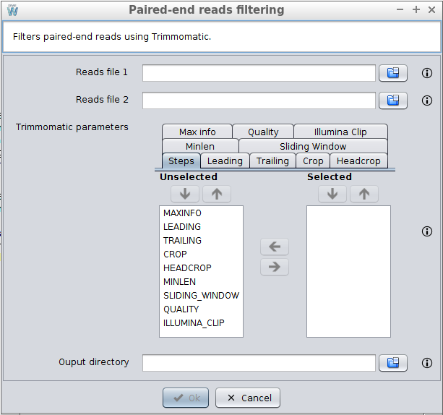
3.1.2.2 Paired-end reads filtering
This operation allows filtering single-end raw reads using Trimmomatic. Clicking on the Trimmomatic (paired-end) button in the Workflow catalog, a new window will be displayed and the following data will be requested:
- Reads file 1: the first reads file.
- Reads file 2: the second reads file.
- Trimmomatic parameters: the steps for trimmomatic and its configuration. The Steps tab allows selecting which steps must be applied and define the order in which they should be applied. Then, the other tabs allows configuring each step. The following steps are available:
- Leading: removes low quality bases from the beginning. As long as a base has a value below this threshold the base is removed and the next base will be investigated.
- Trailing: removes low quality bases from the end. As long as a base has a value below this threshold the base is removed and the next base (which as trimmomatic is starting from the 3' prime end would be base preceding the just removed base) will be investigated. This approach can be used removing the special illumina 'low quality segment' regions (which are marked with quality score of 2), but we recommend Sliding Window or MaxInfo instead.
- Crop: removes bases regardless of quality from the end of the read, so that the read has maximally the specified length after this step has been performed. Steps performed after CROP might of course further shorten the read.
- Headcrop: removes the specified number of bases, regardless of quality, from the beginning of the read.
- Minlen: removes reads that fall below the specified minimal length. If required, it should normally be after all other processing steps. Reads removed by this step will be counted and included in the „dropped reads‟ count presented in the trimmomatic summary.
- Sliding window: performs a sliding window trimming, cutting once the average quality within the window falls below a threshold. By considering multiple bases, a single poor quality base will not cause the removal of high quality data later in the read.
- Max info: performs an adaptive quality trim, balancing the benefits of retaining longer reads against the costs of retaining bases with errors.
- Quality: reencodes the quality part of the FASTQ file to the selected base.
- Illumina clip: finds and removes Illumina adapters.
- Output directory: optionally, the directory where the filtered file must be created. If not provided, the output file is created in the same directory as the reads file being filtered.
3.2 Bowtie2, StringTie and R packages (Ballgown and edgeR)
This workflow was introduced by Griffith, M. et al. [5]. As the title suggests, this workflow makes use of the tools Bowtie2 [6] to align sample reads, StringTie [3] to assemble transcripts and two R packages, Ballgown [4] and EdgeR [7], to perform the differential expression itself:
- Bowtie2 aligns RNA-Seq reads to a genome. This aligner is less exigent than HISAT2 from a computational point of view and can be more suitable for comparisons including smaller reference genomes. However, Bowtie2 was mainly designed to align samples without intron-sized gaps.
- StringTie assembles the alignments into full and partial transcripts, creating multiple isoforms as necessary and estimating the expression levels of all genes and transcripts. StringTie normalises the sequence depth and gene length by reporting the quantification results in FPKM (Fragments Per Kilobase Million) and in TPM (Transcripts Per kilobase Million).
- Ballgown takes the transcripts and expression levels from StringTie normalised in FPKM and applies rigorous statistical methods to determine which transcripts are differentially expressed between the conditions. Besides, edgeR uses raw count produced by HtSeq and then normalises this raw counts in TMM (Trimmed Mean of M-values). A detailed explanation between how the different normalization metrics work, can be found in this excellent online resource: https://www.rna-seqblog.com/rpkm-fpkm-and-tpm-clearly-explained/
Table 2: Condition of each sample used in the workflow.
Step 1: Download the dataset
The example dataset is available at the following URL: http://static.sing-group.org/software/DEWE/data/tutorial_data_HCC1395.zip. This dataset must be downloaded and uncompressed in the application shared folder.
The dataset contains the following files and directories:
- genes: a directory containing the reference annotation file called Homo_sapiens.GRCh38.86.chromosome22.gtf.
- genome: a directory containing the reference genome in FASTQ format.
- indexes: a directory containing the Bowtie2 indexes of the chromosome 22 reference genome.
- samples: a directory containing two folders:
- normal: a directory containing the paired-end reads corresponding to the 3 normal samples in the dataset.
- tumor: a directory containing the paired-end reads corresponding to the 3 tumor samples in the dataset.
Step 2: Configure the workflow
The next step consists in configuring the workflow. To do so, go to the Workflow catalog and click the Run workflow button next to the Bowtie2, StringTie and R workflow description.


Step 3: Import the reference genome index
After clicking the Import index button, the following dialog will appear, allowing to select the downloaded reference genome index. The following data will be requested:
- Index folder: the directory that contains the Bowtie2 genome index. Select the indexes folder in the case study data. When selecting the folder, the files it contains will not be displayed.
- Name: the name for the genome index in order to identify it later.


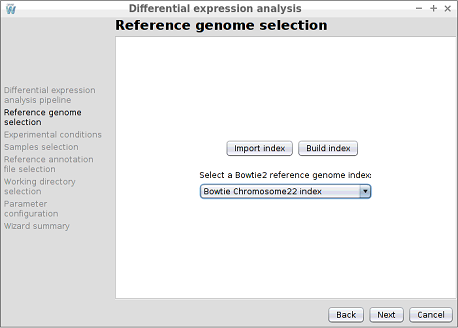
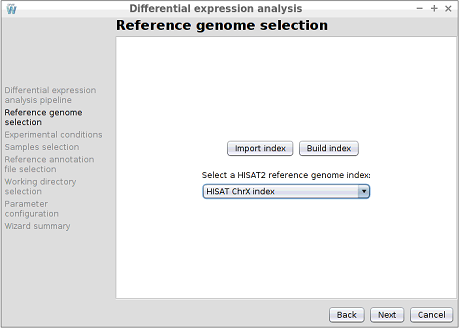
Step 4: Reference genome selection
In this step, the reference genome to be used to perform the alignment must be chosen. As shown in the following image, the configuration assistant shows the available Bowtie2 indexes. Note that in this step you can also use the Import index and Build index buttons to import or create a new reference genome index.

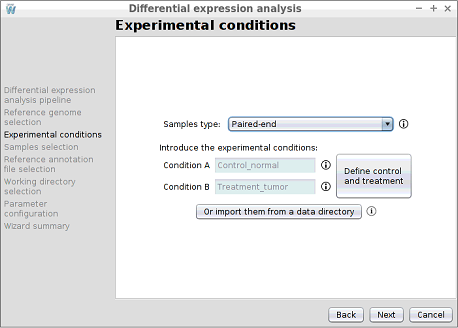
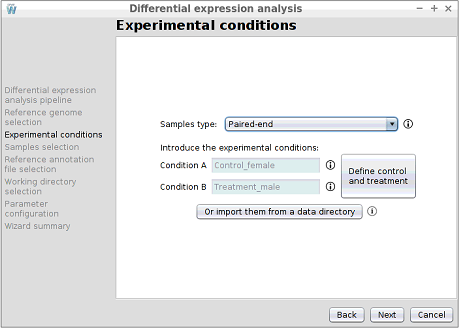
Step 5: Introducing the experimental conditions
In this step, the names of the two experimental conditions of the experiment must be introduced.

- It must contain two folders and the name of each folder will correspond to a condition. In this case, the samples folder contains the normal and tumor folders.
- Each of the two folders must contain the pairwise files of the samples and these files must be in .fq, .fastq or .fastq.gz format. In addition, as samples are paired-end, the first reads file must end in _1 and the second reads in _2.

Advanced: Define control and treatment condition
DEWE determines the control condition as the first alphabetical ordered regardless of the order in which they were entered. To change this, there is a button at the right of the conditions text boxes, Define control and treatment, which opens a new window where conditions can be defined as Control and Treatment.
Step 6: samples verification [Optional: samples selection]
If the samples directory was selected previously, this step can be used to verify that all samples have been selected correctly.

Optional: Samples selection
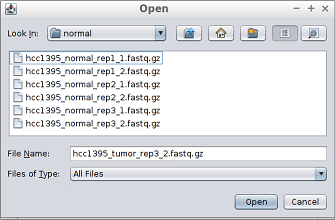
If a samples directory has not been selected in the previous step, the samples must be entered manually:
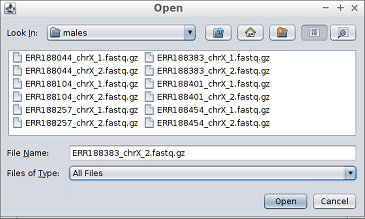
1. Select the first reads file of each sample (those ending with _1.fastq.gz in the samples directory) by clicking the button.
- Reads are provided in compressed FASTQ format so if the file browser is empty when browsing into the samples directory, then the appropriate file filter must be select as the image below shows.
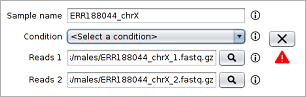
- As can be seen in the image below, after selecting the first reads file the second reads file and the sample name are automatically filled by the tool.
- By default, the list contains four samples. Since in this case you must introduce up to twelve samples, the button must be clicked to introduce more samples.
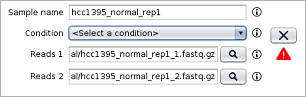
- hcc1395_normal_rep1: normal
- hcc1395_normal_rep2: normal
- hcc1395_normal_rep3: normal
- hcc1395_tumor_rep1: tumor
- hcc1395_tumor_rep2: tumor
- hcc1395_tumor_rep3: tumor
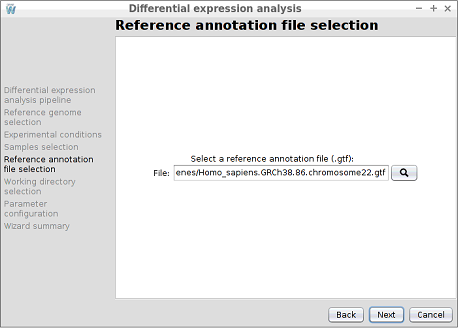
Step 7: Reference annotation file selection
Select the reference annotation file for the experiment. In this case, the Homo_sapiens.GRCh38.86.chromosome22.gtf file located in the genes directory must be selected. After selecting it, the Next button must be clicked to advance to the next step.
A ready-to-use reference sequence list and their annotations can be downloaded form the Illumina iGenome site (https://support.illumina.com/sequencing/sequencing_software/igenome.html), taking into account that only eukaryotic organisms can be analysed through DEWE.
Step 8: Working directory selection
Choose the working directory, where the analysis results will be stored, and advance to the final step.
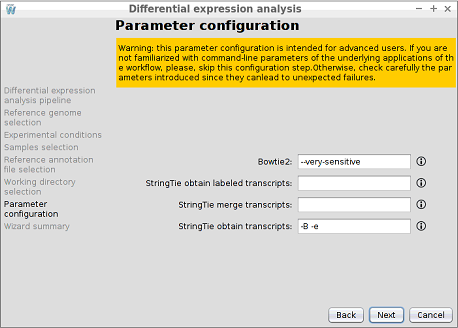
Step 9: Parameter configuration
WARNING: this parameter configuration is intended for advanced users. If you are not familiarized with command-line parameters of the underlying applications of the workflow, please, skip this configuration step. Otherwise, check carefully the parameters introduced since they can lead to unexpected failures.
DEWE allows the user to manually introduce additional parameters to the alignment and transcript reconstruction steps, like a command line execution.
- Bowtie2: custom command-line parameters for the execution of the Bowtie2 alignment. For more information on Bowtie2 options, please, check the reference manual (http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml#options).
- StringTie obtain labeled transcripts: custom command-line parameters for the execution of StringTie reconstruct labeled transcripts command. For more information on StrinTie options, please, check the reference manual (http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual).
- StringTie merge transcripts: custom command-line parameters for the execution of the StringTie merge command. For more information on StrinTie options, please, check the reference manual (http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual).
- StringTie obtain transcripts: custom command-line parameters for the execution of StringTie reconstruct transcripts command. For more information on StrinTie options, please, check the reference manual (http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual).
Step 10: Workflow configuration summary
In this last step a summary of the workflow configuration is provided. It must be checked carefully in order to ensure that the right files were selected, namely the samples’ conditions. If the workflow is executed, this summary is also stored in the selected working directory as workflow-description.txt.

Step 11: Monitoring the workflow execution
While the workflow is being executed, the execution can be monitored in a dialog as the one shown in the following image.

Step 12: Workflow results
The following files and directories are generated by the application in the selected working directory:
- workflow-description.txt: a plain text file containing the workflow configuration (input files, experiment description, etc.).
- run.log: a log containing the executed commands.
- read-mapping-statistics.csv: a table containing the statistics of the alignment for each sample.
- stringtie: a directory containing the StringTie merged annotation file.
- Directories for each of the samples: each sample’s directory contains the files produced by the workflow components such as the alignments (in .sam and .bam formats) and the transcripts calculated by StringTie.
- analysis: a directory containing the following subdirectories with the results of each package:
- analysis/ballgown: this directory contains the results of the differential expression analysis performed with Ballgown. See subsection 4.1 for more information on the generated outputs.
- analysis/edger: this directory contains the results of the differential expression analysis performed with edgeR. See subsection 4.2 for more information on the generated outputs.
- analysis/overlaps: this directory contains the summary of the overlapped significantly differentially expressed genes between Ballgown and edgeR analyses. See subsection 4.3 for more information on the generated outputs.
3.3 HISAT2, StringTie and Ballgown
This workflow was introduced by Pertea, M. et al. [1]. As the title suggests, this workflow mekes use of the tools HISAT2 [2] to align sample reads, StringTie [3] to assemble transcripts, and Ballgown [4] to perform the differential expression analysis:
- HISAT2 aligns RNA-Seq reads to a genome and discovers transcript splice sites. This aligner is more exigent than Bowtie2 from a computational point of view but more accurate. Moreover, HISAT2 is superior to Bowtie2 in aligning across intron-sized gaps.
- StringTie assembles the alignments into full and partial transcripts, creating multiple isoforms as necessary and estimating the expression levels of all genes and transcripts. StringTie normalises the sequence depth and gene length by reporting the quantification results in FPKM (Fragments Per Kilobase Million) and in TPM (Transcripts Per kilobase Million).
- Ballgown takes the transcripts and expression levels from StringTie normalised in FPKM and applies rigorous statistical methods to determine which transcripts are differentially expressed between the conditions. Besides, edgeR uses raw count produced by HtSeq and then normalises this raw counts in TMM (Trimmed Mean of M-values). A detailed explanation between how the different normalization metrics work, can be found in this excellent online resource: https://www.rna-seqblog.com/rpkm-fpkm-and-tpm-clearly-explained/
Table 3: Population and sex of each sample used in the workflow.
Step 1: Download the dataset
The example dataset is available at the following URL: http://static.sing-group.org/software/DEWE/data/tutorial_data_chrX.zip. This dataset must be downloaded and uncompressed in the application shared folder.
The dataset contains the following files and directories:
- genes: a directory containing the reference annotation file called chrX.gtf.
- genome: a directory containing the reference genome in FASTQ format.
- indexes: a directory containing the HISAT2 indexes of the chromosome X reference genome.
- samples: a directory containing two folders:
- females: a directory containing the paired-end reads corresponding to the 6 female samples in the dataset.
- males: a directory containing the paired-end reads corresponding to the 6 male samples in the dataset.
- geuvadis_phenodata.csv: a CSV file that contains the phenotype or condition of each sample. For this tutorial, it is important the classification of samples in male and female.
Step 2: Configure the workflow
The next step consists in configuring the workflow. To do so, go to the Workflow catalog and click the Run workflow button next to the HISAT2, StringTie and Ballgown workflow description.

When the workflow is executed for the first time or no HISAT2 reference genome indexes are available, the tool requires the importation or creation of a reference genome index using HISAT2.

Step 3: Import the reference genome index
After clicking the Import index button, the following dialog will appear, allowing to select the downloaded reference genome index. The following data will be requested:
- Index folder: the directory that contains the HISAT2 genome index. Select the indexes folder in the case study data. When selecting the folder, the files it contains will not be displayed.
- Name: the name for the genome index in order to identify it later.


Step 4: Reference genome selection
In this step, the reference genome to be used to perform the alignment must be chosen. As shown in the following image, the configuration assistant shows the available HISAT2 indexes. Note that in this step you can also use the Import index and Build index buttons to import or create a new reference genome index.

Step 5: Introducing the experimental conditions
In this step, the names of the two experimental conditions of the experiment must be introduced.
- It must contain two folders and the name of each folder will correspond to a condition. In this case, the samples folder contains the females and males folders.
- Each of the two folders must contain the pairwise files of the samples and these files must be in .fq, .fastq or .fastq.gz format. In addition, as samples are paired-end, the first reads file must end in _1 and the second reads in _2.

Advanced: Define control and treatment condition
DEWE determines the control condition as the first alphabetical ordered regardless of the order in which they were entered. To change this, there is a button at the right of the conditions text boxes, Define control and treatment, which opens a new window where conditions can be defined as Control and Treatment.
Step 6: samples verification [Optional: samples selection]
If the samples directory was selected previously, this step can be used to verify that all samples have been selected correctly.

Optional: Samples selection
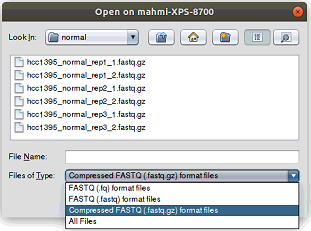
If a samples directory has not been selected in the previous step, the samples must be entered manually:1. Select the first reads file of each sample (those ending with _1.fastq.gz in the samples directory) by clicking the button.
- Reads are provided in compressed FASTQ format so if the file browser is empty when browsing into the samples directory, then the appropriate file filter must be select as the image below shows.
- As can be seen in the image below, after selecting the first reads file the second reads file and the sample name are automatically filled by the tool.
- By default, the list contains four samples. Since in this case you must introduce up to twelve samples, the button must be clicked to introduce more samples.
- ERR188044: male
- ERR188104: male
- ERR188234: female
- ERR188245: female
- ERR188257: male
- ERR188273: female
- ERR188337: female
- ERR188383: male
- ERR188401: male
- ERR188428: female
- ERR188454: male
- ERR204916: female
Step 7: Reference annotation file selection
Select the reference annotation file for the experiment. In this case, the chrX.gtf file located in the genes directory must be selected. After selecting it, the Next button must be clicked to advance to the next step.

A ready-to-use reference sequence list and their annotations can be downloaded form the Illumina iGenome site (https://support.illumina.com/sequencing/sequencing_software/igenome.html), taking into account that only eukaryotic organisms can be analysed through DEWE.
Step 8: Working directory selection
Choose the working directory, where the analysis results will be stored, and advance to the final step.
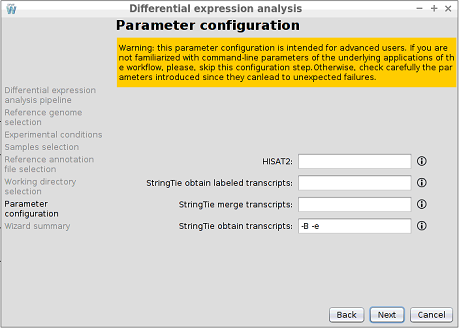
Step 9: Parameter configuration
WARNING: this parameter configuration is intended for advanced users. If you are not familiarized with command-line parameters of the underlying applications of the workflow, please, skip this configuration step. Otherwise, check carefully the parameters introduced since they can lead to unexpected failures.
DEWE allows the user to manually introduce additional parameters to the alignment and transcript reconstruction steps, like a command line execution.
- HISAT2: custom command-line parameters for the execution of the HISAT2 alignment. For more information on HISAT2 options, please, check the reference manual (https://ccb.jhu.edu/software/hisat2/manual.shtml#command-line-1).
- StringTie obtain labeled transcripts: custom command-line parameters for the execution of StringTie reconstruct labeled transcripts command. For more information on StrinTie options, please, check the reference manual (http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual).
- StringTie merge transcripts: custom command-line parameters for the execution of the StringTie merge command. For more information on StrinTie options, please, check the reference manual (http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual).
- StringTie obtain transcripts: custom command-line parameters for the execution of StringTie reconstruct transcripts command. For more information on StrinTie options, please, check the reference manual (http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual).
Step 10: Workflow configuration summary
In this last step a summary of the workflow configuration is provided. It must be checked carefully in order to ensure that the right files were selected, namely the samples’ conditions. If the workflow is executed, this summary is also stored in the selected working directory as workflow-description.txt.

Step 11: Monitoring the workflow execution
While the workflow is being executed, the execution can be monitored in a dialog as the one shown in the following image.

Step 12: Workflow results
The following files and directories are generated by the application in the selected working directory:
- workflow-description.txt: a plain text file containing the workflow configuration (input files, experiment description, etc.).
- run.log: a log containing the executed commands.
- read-mapping-statistics.csv: a table containing the statistics of the alignment for each sample.
- stringtie: a directory containing the StringTie merged annotation file.
- Directories for each of the samples: each sample’s directory contains the files produced by the workflow components such as the alignments (in .sam and .bam formats) and the transcripts calculated by StringTie.
- analysis: a directory containing the following subdirectories with the results of each package:
- analysis/ballgown: this directory contains the results of the differential expression analysis performed with Ballgown. See subsection 4.1 for more information on the generated outputs.
- analysis/edger: this directory contains the results of the differential expression analysis performed with edgeR. See subsection 4.2 for more information on the generated outputs.
- analysis/overlaps: this directory contains the summary of the overlapped significantly differentially expressed genes between Ballgown and edgeR analyses. See subsection 4.3 for more information on the generated outputs.
3.4 Configure a workflow using the workflow.dewe file
When configuring a workflow, DEWE has two methods. On the one hand, following the steps one by one of sections 3.2 and 3.3 and manually introducing each input. On the other hand, through a workflow configuration file named workflow.dewe that automatically introduces all inputs.
This configuration file contains information related to inputs and configuration of the workflow, i.e. the location of the input files (read files of the samples and annotation file), the two conditions, the index selected for the alignment and the output folder. When the configuration file is uploaded, DEWE will fill in all the inputs, but allowing its inspection and modification. A workflow.dewe file is generated on the output folder each time an analysis is executed. This file can not be edited manually.
The main advantage of this method is that it allows the re-execution of an analysis quickly. For example, if the user runs an analysis from one workflow and wants to run it in the other, just need to upload the configuration file. Another interesting example would be re-execute an analysis, but changing a single parameter (e.g., remove a sample).
To execute a workflow from a configuration file, in the workflows catalog, click on the "Import workflow" button, which will open a file browser to select the configuration file to be imported.

3.5 Workflow execution FAQ
3.5.1 Error executing bowtie2-build

3.5.2 No reads files are displayed in the sample selection
When selecting the sample files manually, the directory where the samples are found may appear empty.

3.5.3 java.io.IOException error during alignment

3.5.3.1 Could not locate a Bowtie/HISAT index
Could not locate a [Bowtie/HISAT] index corresponding to basename "/mnt/shared/mahmi/rna_seq/sf_data/Data/test/indexes/Homo_sapiens.GRCh38.dna_sm.chromosome.22index"
In the sample.sam.txt file
This is because the index was imported / built correctly but later its location has been deleted or changed. The user must import / build the index again.
3.5.3.2 Reads file does not look like a FASTQ file
Error: reads file does not look like a FASTQ file
In the sample.sam.txt file
This is because some of the reading files are wrong. The user must verify the sample files for errors.
3.5.4 Error executing StringTie

3.5.5 Workflow execution error

3.5.6 Invalid workflow file

3.5.7 Heatmap and PCA plot have not been generated after Ballgown execution
This happens when an analysis does not produce genes with a q-value < 0.05. If in an analysis these figures are not generated, it means that statistically significant genes have not been found.
Table of contents
- 4. Outputs and visualisation
- 4.1 Ballgown
- 4.1.1 Ballgown outputs
- 4.1.2 Results visualisation
- 4.1.2.1 Creation of additional results from transcripts tables
- 4.1.2.2 Creation of additional results from genes tables
- 4.1.2.3 Creation of additional filtered genes tables
- 4.1.2.4 Creation of additional filtered transcripts tables
- 4.1.2.5 Creation of colored figures
- 4.1.2.6 Visualisation of the additional filtered tables
- 4.2 edgeR
- 4.3 Overlaps between Ballgown and edgeR analyses
4. Outputs and visualisation
4.1 Ballgown
The Ballgown differential expression analysis can be run from the menu operation (PDF section 5.7.1) or as part of the available workflows (sections 3.2 and 3.3). The outputs are similar in both cases. This section explains these outputs and the visualisation capabilities of the tool.
4.1.1 Ballgown outputs
After performing a differential expression analysis with Ballgown, the following single analysis are generated:
- consensuspathdb_enrichment_analysis.csv: input file for ConsensusPathDB gene set enrichment analysis.
- phenotype-data_gene_results.tsv: differentially expressed genes between the two conditions.
- phenotype-data_gene_results_filtered.tsv: differentially expressed genes between the two conditions, low-abundance genes have been filtered. The transcripts with variance across the samples of less than one have been removed.
- phenotype-data_gene_results_sig.tsv: differentially expressed genes between the two conditions, with p-value less than 5%.
- phenotype-data_transcript_results.tsv: differentially expressed transcripts between the two conditions.
- phenotype-data_transcript_results_filtered.tsv: differentially expressed transcripts between the two conditions, low-abundance genes have been filtered. The transcripts with variance across the samples of less than one have been removed.
- phenotype-data_transcript_results_sig.tsv: differentially expressed transcripts between the two conditions, with p-value less than 5%.
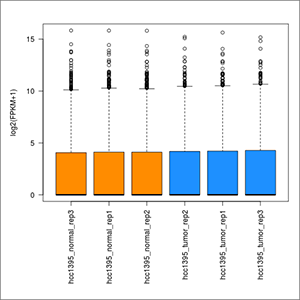
- FPKM-distribution-across-samples.jpeg: plot of the distribution of FPKM values across the samples. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
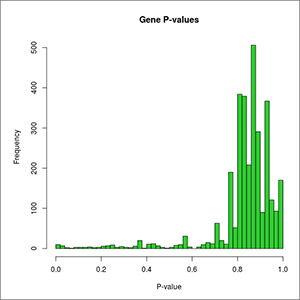
- genes-DE-pValues-distribution.jpeg: plot of the overall distribution of differential expression p-values for genes. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
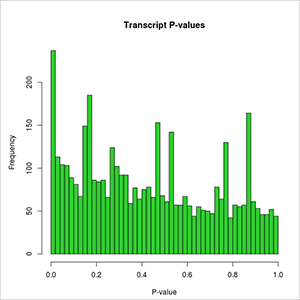
- transcripts-DE-pValues-distribution.jpeg: plot of the overall distribution of differential expression p-values for transcripts. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
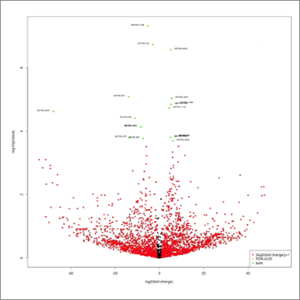
- volcano-plot.jpeg: This graphic combines the results of the p-values obtained after statistical tests with every single fold change. It will allow a rapid identification of the genes that increases both their magnitude of fold change and their statistical significance. Orange color represent up- or down-regulated genes (absolute value of log2fold-change > 1). Red color represent genes changing their expression with a statistical significance equal or lower than 0.05, measured as the adjusted p-value using the false discovery rate method. Green color represent up- or down-regulated genes with combine the two previous criteria. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
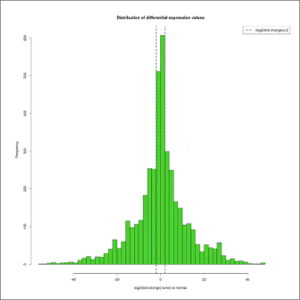
- DE-values-distribution.jpeg: distribution of differential expression values histogram, which shows the frequency of the different expression fold-changes in a given experiment. This will give the user an idea of the proportion of genes changing their expression over a given fold-change threshold, which is depicted with a dashed line. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
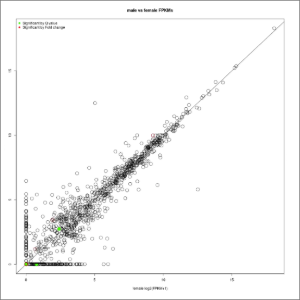
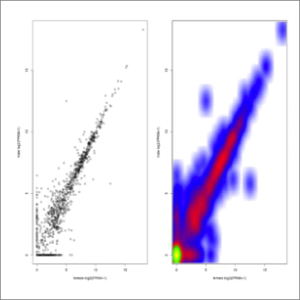
- FPKM-conditions-correlation.jpeg: FPKMs correlation between conditions, which are plotted in the x/y axis and will highlight those genes with enriched FPKM values in one or other experimental condition. Genes showing no variation in their experimental FPKM values will adjust to the diagonal line. Note that values are expressed in FPKM+1 values, avoiding dividing by zero when calculating fold changes. Red color represent up- or down-regulated genes (absolute value of log2fold-change > 2). Red color represent genes changing their expression with a statistical significance equal or lower than 0.05, measured as the adjusted p-value using the false discovery rate method. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
- FPKM-conditions-density.jpeg: FPKM correlation between conditions, represented as a density plot. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
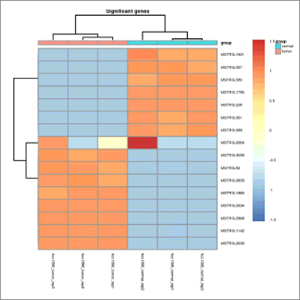
- Heatmap.jpeg: Heatmap of statistically significant genes (q-value<0.05), which a computed row and column clustering, the latter with an additional layer including the phenotype data. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
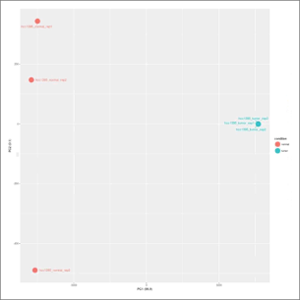
- pca.jpeg: Principal Component Analysis plot, in which the global variance of the experiment is decomposed and grouped into new and orthogonal variables denominated components. PCA allows a quick inspection on the factors driving the observed variance, including unsupervised discovery of confounding factors. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the Ballgown results folder.
- user-images/FPKM-distribution-gene_genename-transcript_transcriptname.format: FPKM distributions for a given transcript. This image can be exported in jpeg, tiff or png and in the resolution and color (colored or grayscale) defined by the user.
- user-images/transcripts-gene_genename-sample_samplename.format: Structure and expression levels of the distinct isoform for a given transcript gene in a given sample. This image can be exported in jpeg, tiff or png and in the resolution and color (colored or grayscale) defined by the user.
- user-tables/transcript_results_sig_percentage.tsv: differentially expressed transcripts between the two conditions, with p-value less than the selected percentage.
- user-tables/gene_results_sig_percentage.tsv: differentially expressed genes between the two conditions, with p-value less than the selected percentage.
- A list, named by the user, with the selected number of the most overexpressed or underexpressed genes. This is an input for ConsensusPathDB over-representation gene set analysis.
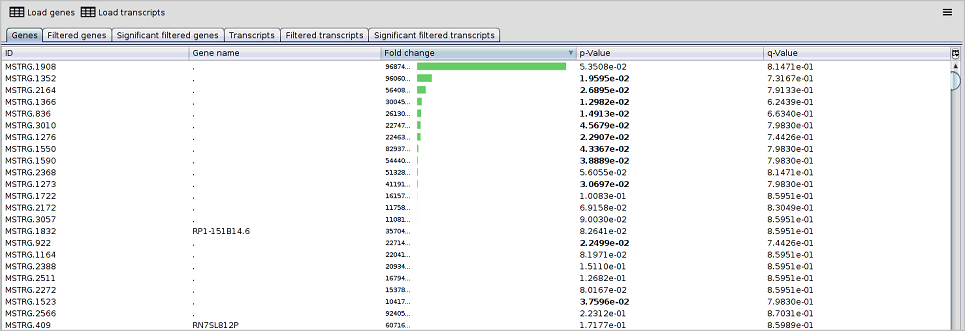
4.1.2 Results visualisation
The viewer enables the interactive browsing of genes and transcripts analysis through some generated tables to better understand and visualisation this analysis in the Ballgown working directory.
As you be seen in the following image, this view contains the following six tabs:
- Genes: this tab contains a table with the genes in file phenotype-data_gene_results.tsv.
- Filtered genes: this tab contains a table with the genes in file phenotype-data_gene_results_filtered.tsv.
- Significant filtered genes: this tab contains a table with the genes in file phenotype-data_gene_results_sig.tsv.
- Transcripts: this tab contains a table with the transcripts in file phenotype-data_transcript_results.tsv.
- Filtered transcripts: this tab contains a table with the transcripts in file phenotype-data_transcript_results_filtered.tsv.
- Significant filtered transcripts: this tab contains a table with the transcripts in file phenotype-data_transcript_results_sig.tsv.
4.1.2.1 Creation of additional results from transcripts tables
Any of the transcripts tables allows to generate the first two additional analysis described in section 4.1.1. To perform this analysis, first the rows corresponding to the transcripts that are wanted to export must be selected. Then, right-click must be done in order to make visible a contextual menu with options to create the figures that explain these analysis.


4.1.2.2 Creation of additional results from genes tables
Any of the gene tables allows to generate the last additional analysis described in section 4.1.1. To perform this analysis, first the button over the vertical scroll must be clicked. Then, the Export gene names option must be selected. This will open a new window.

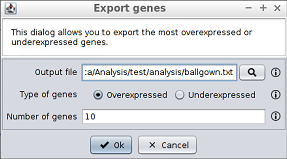
4.1.2.3 Creation of additional filtered genes tables
The Filtered genes table allows to generate the third analysis described in section 4.1.1. To perform this analysis, first the button over the vertical scroll must be clicked. Then, the Filter and export genes option must be selected. This will open a new window.


4.1.2.4 Creation of additional filtered transcripts tables
The Filtered transcripts table allows to generate the fourth analysis described in section 4.1.1. To perform this analysis, first the button over the vertical scroll must be clicked. Then, the Filter and export transcripts option must be selected. This will open a new window.


4.1.2.5 Creation of colored figures
Over the tabs of the different tables generated, left aligned, there are three buttons that allow you to regenerate the following figures again:
- FPKM across samples: plot of the distribution of FPKM values across the samples.
- genes DE pValues distribution: plot of the overall distribution of differential expression p-values for genes.
- transcripts DE pValues distribution: plot of the overall distribution of differential expression p-values for transcripts
- Volcano plot: combines the results of the p-values obtained after statistical tests with every single fold change.
- Fold changes DE values distribution: distribution of differential expression values histogram, which shows the frequency of the different expression fold-changes in a given experiment.
- FPKM correlation plot: FPKMs correlation between conditions, which are plotted in the x/y axis and will highlight those genes with enriched FPKM values in one or other experimental condition.
- FPKM density plot: FPKM correlation between conditions, represented as a density plot.
- Heatmap: heatmap of statistically significant genes (q-value<0.05), which a computed row and column clustering, the latter with an additional layer including the phenotype data.
- Principal Component Analysis: Principal Component Analysis: the global variance of the experiment is decomposed and grouped into new and orthogonal variables denominated components.

4.1.2.6 Visualisation of the additional filtered tables
Over the tabs of the different tables generated, right aligned, there are two buttons that allow you to load the previous generated additional filter tables for genes and transcripts:
- Load genes: Load a user-created filtered genes table.
- Load transcripts: Load a user-created filtered transcripts table.


4.2 edgeR
The edgeR differential expression analysis can be run from the menu operation (PDF section 5.7.2) or as part of the workflows (sections 3.2 and 3.3). This section explains these outputs and the visualisation capabilities of the tool.
4.2.1 edgeR outputs
After performing a differential expression analysis with edgeR, the following single analysis are generated:
- DE_genes.txt: differentially expressed genes between the two conditions.
- DE_significant_genes.tsv: significant differentially expressed genes between the two conditions, i.e. genes with p-value<0.05.
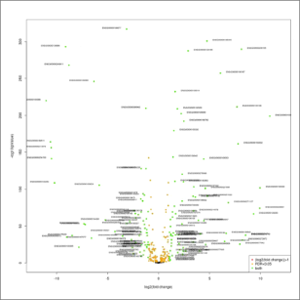
- volcano-plot.jpeg: This graphic combines the results of the p-values obtained after statistical tests with every single fold change. It will allow a rapid identification of the genes that increases both their magnitude of fold change and their statistical significance. Orange color represent up- or down-regulated genes (absolute value of log2fold-change > 1). Red color represent genes changing their expression with a statistical significance equal or lower than 0.05, measured as the adjusted p-value using the false discovery rate method. Green color represent up- or down-regulated genes with combine the two previous criteria. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the edgeR results folder.
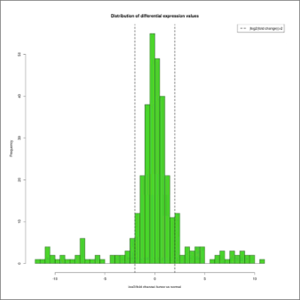
- DE-values-distribution.jpeg: distribution of differential expression values histogram, which shows the frequency of the different expression fold-changes in a given experiment. This will give the user an idea of the proportion of genes changing their expression over a given fold-change threshold, which is depicted with a dashed line. At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the edgeR results folder.
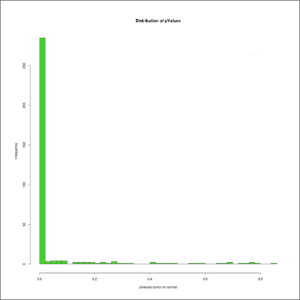
- pValues-distribution.jpeg: histogram of the frequencies of experimental p-values corresponding to the genes contained in the analysed genome. It will provide the user with a distribution of the differential expression p-values for all the genes, and it will also provide an indication of the subset of genes without significative differences in their fold-changes (p-value > 0.05). At first this image will be created in grayscale, but later it will be able to be generated again in the format, size and color chosen by the user (colored or grayscale). These user generated images will be saved in the user-images folder, contained within the edgeR results folder.
4.2.2 Results visualisation
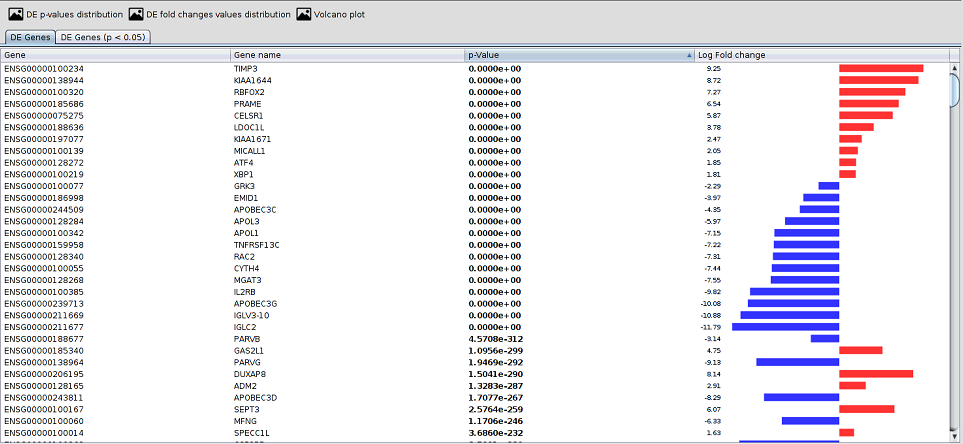
The viewer enables the interactive browsing of genes analysis through a generated table to better understand and visualisation this analysis in the edgeR working directory.
As you can see in the following image, this view contains two tabs:
- DE Genes: this tab contains a table with the genes in file DE_genes.tsv
- DE Genes (p<0.05): this tab contains a table with the genes in file DE_significant_genes.tsv
4.2.2.1 Creation of colored figures
Over the tabs of the different tables generated, left aligned, there are three buttons that allow you to regenerate the following figures again:
- genes DE p-values distribution: plot of the overall distribution of differential expression p-values for genes.
- Fold changes DE values distribution: distribution of differential expression values histogram, which shows the frequency of the different expression fold-changes in a given experiment.
- Volcano plot: combines the results of the p-values obtained after statistical tests with every single fold change.

4.3 Overlaps between Ballgown and edgeR analyses
After the execution of the workflows, a summary of the overlaps between the significantly differentially expressed genes (q-value<0.05) of Ballgown and edgeR analysis will be provided.
4.3.1 edgeR outputs
After performing the differential expression analysis with Ballgown and edgeR, the following single analysis are generated:
- overlap-ballgown-edger.tsv: a table containing the common significantly DE genes (q-value<0.05) between Ballgown and edgeR analyses.
- overlap_ballgown_edger.tif: a Venn diagram summarising the overlap between Ballgown and edgeR analyses.
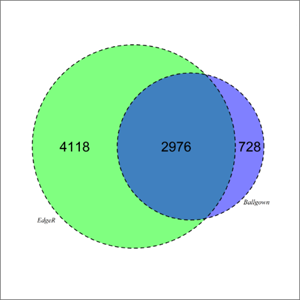
4.3.2 Results visualisation
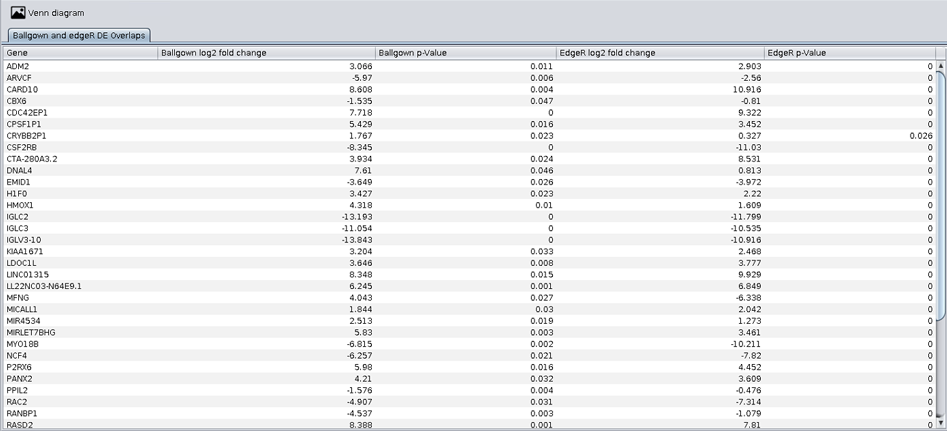
The viewer enables the interactive browsing of the overlapping genes through a generated table to better understand and visualisation this analysis in the overlapping working directory.
As you can see in the following image, this view contains one tab:
- Ballgown and edgeR DE overlaps: this tab contains a table with the overlapping genes in file overlap-ballgown-edger.tsv.
4.3.2.1 Creation of colored figures
Over the tabs of the table generated, left aligned, there are one button that allow you to regenerate the following figure again:
- Venn diagram: Venn diagram summarising the overlap between Ballgown and edgeR analyses.

Table of contents
5. Case study
DEWE may be an appropriate tool for translational studies, notably those of clinical or biotechnological interest. In order to evidence this translational application, this section presents a case study of biomedical interest where monocytes and monocyte-derived dendritic cells (moDCs) are compared.
5.1 Introduction
Monocytes are essential elements in the immune response against pathogens, and inflammatory processes caused by external aggressions, infection and autoimmune disease [8]. Moreover, monocytes have been proved to be endowed with the potential to differentiate into dendritic cells (DCs) or macrophages in vivo during inflammation [9].

Given the scarcity of peripheral blood DCs and the ethical and technical difficulties involved in obtaining tissue-derived DCs from human sources, investigators have resorted to using different model systems for studying DC biology [15]. For instance, DCs can be generated in vitro from peripheral blood monocytes by culturing them for six days in the presence of interleukin (IL)-4 and granulocyte-macrophage colony stimulating factor (GM-CSF). Under such culture conditions, cells acquire an immature DC morphology and express DC differentiation antigens [16]. These moDCs are routinely used as an mDC model in DC research.
Previous studies have conducted transcriptome comparative analyses between monocytes and monocyte-derived macrophages in order to better understand the molecular mechanisms involved in monocyte to macrophage differentiation [17], but to our knowledge, a transcriptome analysis between monocytes and moDC has not yet been performed.
5.2 Results and Discussion
From 4 different samples of monocytes and moDCs, using the “HISAT2, StringTie and R packages” workflow, a differential expression analysis was executed (the dataset is available at the following URL: http://static.sing-group.org/software/DEWE/data/monocytes_moDCs.zip). Results obtained in this analysis reveal that, as in the monocyte-derived macrophage case, genes encoding transcription regulatory factors, such as FOXO1, RUNX3 and C/EBPo were highly expressed in monocytes. Comparing the top 10 up/down-regulated genes of the case of monocyte-derived macrophages, some interesting differences were found. In the monocyte-derived macrophages case, the top up-regulated genes, i.e. C1QC, TREM2, HAMP, APOE, PPARG, CEP55, SYK and APOC1, are also over-expressed in the moDCs case. However, the picture changes with the top under-expressed genes. Genes such as SYN1 and SERPINA1 were also under-expressed, but genes such as DNM2, OLR1, S100A12, CRLF2 and PRM8, which are among the top 10 monocyte-derived macrophages down-regulated genes, presented high levels of over-expression, e.g. OLR1 gene obtains a log2-fold change value of 12.14.
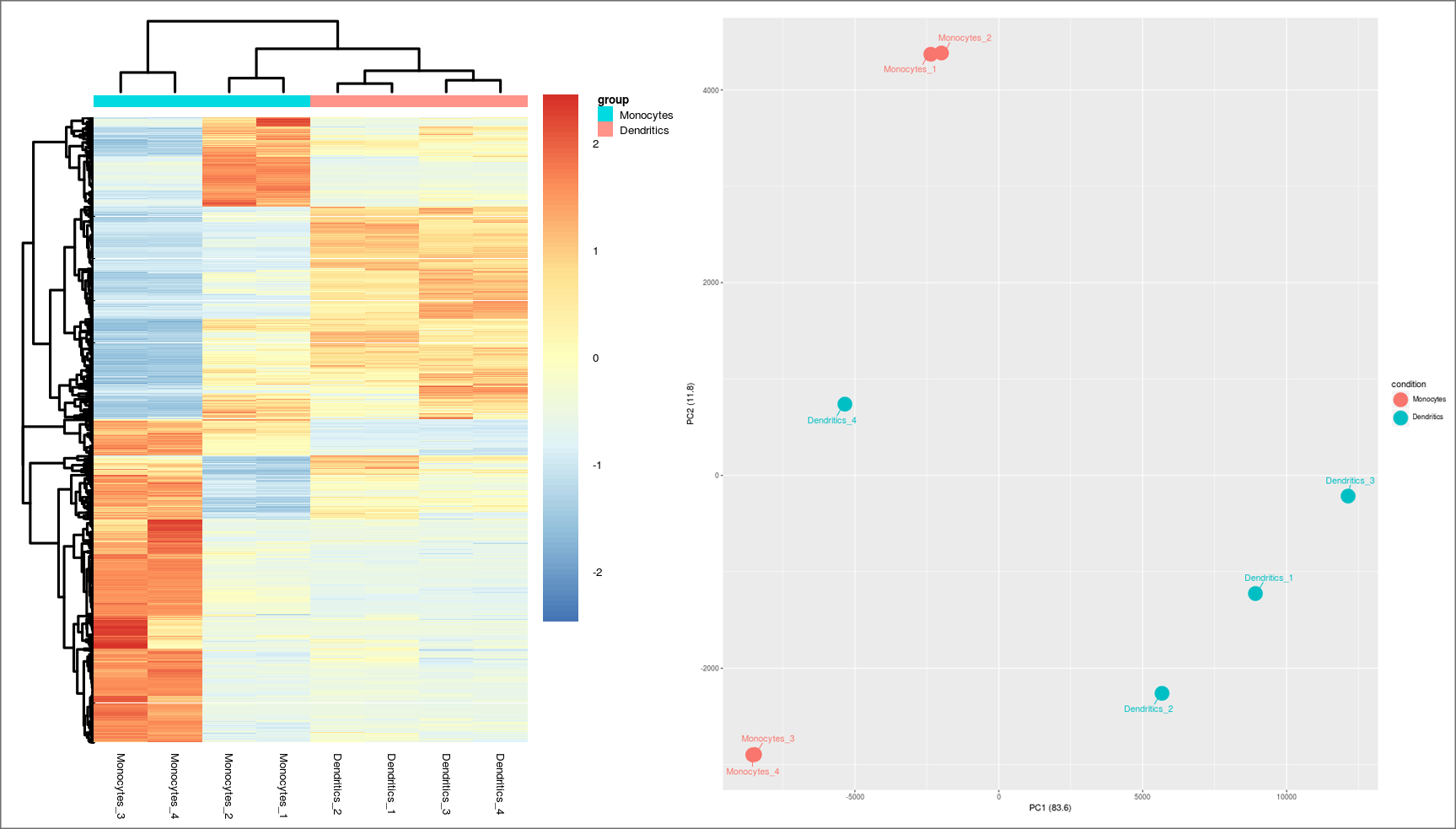
Other interesting expressed gene is CD14. Monocyte cells are known as CD14+ while moDCs are CD14- [18]. This is perfectly shown in the analysis, with a log2-fold change value of -4.06 for this gene. Previous studies demonstrated that mature MoDCs expressed high levels of MHC class II, a protein complex responsible for the presentation of antigens to T cells, and costimulatory molecules [19]. Our experimental RNA-Seq results showed that both HLA-DRB1 and HLA-DRB6 were over-expressed in moDCs. Interestingly, the HLA-DRA1 gene was found under-expressed, which was unexpected as the MHC class II is a heterodimer composed of an alpha chain encoded by HLA-DRA gene and a beta chain encoded by one of the HLA-DRB genes.
This apparent discordance in the regulation might be due to inter-individual variability [20]. However, and although some costimulatory molecules necessary in the moDC-T cell interaction were overexpressed (CD40: log2-fold change value of 8.42 or CD80: log2-fold change value of 4.45) other genes mandatory in this interaction such as CD86, ICOSL, OX40L, PD-L1 and PD-L2 did not increased their expression in our experimental settings. All these genes are mandatory for T cell activation [21]. In this sense, the maturation marker CD83 was not overexpressed in moDCs, so RNA-Seq results supported the idea that moDCs generated in vitro are immature DCs with a limited ability to activate T cells.
6. References
1. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-Seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11: 1650–1667.
2. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12: 357–360.
3. Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat Biotechnol. 2015;33: 290–295.
4. Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33: 243–246.
5. Griffith M, Walker JR, Spies NC, Ainscough BJ, Griffith OL. Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud. PLoS Comput Biol. 2015;11: e1004393.
6. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9: 357–359.
7. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26: 139–140.
8. B. León and C. Ardavín, Monocyte-derived dendritic cells in innate and adaptive immunity, Immunol. Cell Biol., vol. 86, no. 4, pp. 320–324, Jun. 2008.
9. B. León, M. López-Bravo, and C. Ardavín, Monocyte-derived dendritic cells, Semin. Immunol., vol. 17, no. 4, pp. 313–318, Aug. 2005.
10. B. León, M. López-Bravo, and C. Ardavín, Monocyte-Derived Dendritic Cells Formed at the Infection Site Control the Induction of Protective T Helper 1 Responses against Leishmania, Immunity, vol. 26, no. 4, pp. 519–531, Apr. 2007.
11. M. Le Borgne et al., Dendritic Cells Rapidly Recruited into Epithelial Tissues via CCR6/CCL20 Are Responsible for CD8+ T Cell Crosspriming In Vivo, Immunity, vol. 24, no. 2, pp. 191–201, Feb. 2006.
12. H. Tezuka et al., Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells, Nature, vol. 448, no. 7156, pp. 929–933, Aug. 2007.
13. Q. Ji, L. Castelli, and J. M. Goverman, MHC class I–restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8+ T cells, Nat. Immunol., vol. 14, no. 3, pp. 254–261, Mar. 2013.
14. A. Dzionek et al., BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood, J. Immunol., vol. 165, no. 11, pp. 6037–46, Dec. 2000.
15. C. Horlock, F. Shakib, J. Mahdavi, N. S. Jones, H. F. Sewell, and A. M. Ghaemmaghami, Analysis of proteomic profiles and functional properties of human peripheral blood myeloid dendritic cells, monocyte-derived dendritic cells and the dendritic cell-like KG-1 cells reveals distinct characteristics, Genome Biol., vol. 8, no. 3, p. R30, 2007.
16. F. Sallusto and A. Lanzavecchia, Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha, J. Exp. Med., vol. 179, no. 4, pp. 1109–18, Apr. 1994.
17. C. Dong, G. Zhao, M. Zhong, Y. Yue, L. Wu, and S. Xiong, RNA sequencing and transcriptomal analysis of human monocyte to macrophage differentiation, Gene, vol. 519, no. 2, pp. 279–287, May 2013.
18. M. Collin and V. Bigley, Human dendritic cell subsets: an update, Immunology, vol. 154, no. 1, pp. 3–20, May 2018.
19. S. J. Sung, Monocyte-Derived Dendritic Cells as Antigen-Presenting Cells in T-Cell Proliferation and Cytokine Production, 2008, pp. 97–106.
20. M. Kitcharoensakkul et al., Temporal biological variability in dendritic cells and regulatory T cells in peripheral blood of healthy adults, J. Immunol. Methods, vol. 431, pp. 63–65, Apr. 2016.
21. M. Hubo et al., Costimulatory Molecules on Immunogenic Versus Tolerogenic Human Dendritic Cells, Front. Immunol. Methods, 4:82, Apr. 2013.