 BLAST DataBase Manager
BLAST DataBase Manager
Use Cases
- Editing large files
- Retrieving the CDS of intronless genes
- Annotating alternative splicing isoforms
- Deriving alternative gene annotations
- Local Blast
- Concatenating sequences for phylogenetic analyses
- Transferring gene annotations between distantly related species
Use Case 1. Editing large files
Suppose that we want to obtain the phylogeny of the genus Bactrocera (the genus to which the pest species Bactrocera invadens belongs to) based on all COI (cytochrome oxidase subunit I) gene sequences available at NCBI.
- The first step is to obtain the multi FASTA file with the relevant sequences:
- Go to http://www.ncbi.nlm.nih.gov/ and Log in.
- Choose the Nucleotide database and writing in the search bar: "Bactrocera"[Organism] AND "COI"[Gene] NOT "Genome". The NOT "Genome" part of the expression will avoid downloading entire mitochondrial genomes.
- Download all sequences choosing the Send to File option, and selecting FASTA as format.
- The resulting 3.4 Mb file1 can be imported to BDBM (using the
File → Import FASTA operation or by directly placing the file
into
/fasta/nucleotidesfolder of the repository) and inspected (double click on the file under the folderFASTA Files).
-
Note that the definition line is long, that there are blank lines between sequences and line
breaks within sequences. These issues may prevent the use of this file by the researchers'
favorite software package.
All these issues can be fixed using the Operations → Reformat Fasta operation.
In this operation you will find the following options:
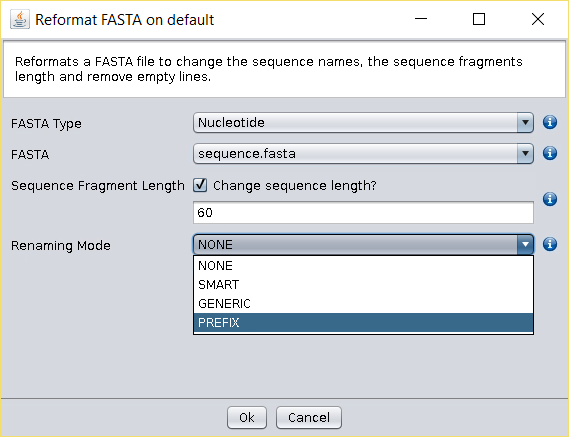
- Sequence Fragment Length: Choosing a negative or zero sequence fragment length will remove all line breaks in the nucleotide sequences, while a positive number, for instance 60, will place a line break every 60 nucleotides (an option that may be useful when preparing FASTA files to be deposited elsewhere).
- Renaming Mode:
- NONE: sequence name will remain unchanged.
- SMART: automatically extracts the gi (general identifier) codes.
- GENERIC: extracts the information present in the fields that are delimited by the | symbol (the first field is 0).
- PREFIX: allows the incorporation of a prefix into all sequence names with the possibility of erasing or keeping the headers.
Final note
These are basic operations but when dealing with large files, such as the one here considered, it may represent a major hurdle for someone without training in informatics.
1 On the 9th of June 2016, 3930 COI sequences could be retrieved.
Use Case 2. Retrieving the CDS of intronless genes
Suppose that we want to prepare a FASTA file to build a phylogeny of the SFBB F-box gene family of Fragaria vesca (strawberry). This may seem a challenging task, since there is no annotation for this species. Nevertheless, this is quite simple using BDBM.
- Download the Fragaria vesca genome FASTA file from here (mirror).
- Extract the FASTA file from the compressed package.
- Import the downloaded Fragaria versca FASTA file to BDBM using the
File → Import FASTA option (or copy the file to the
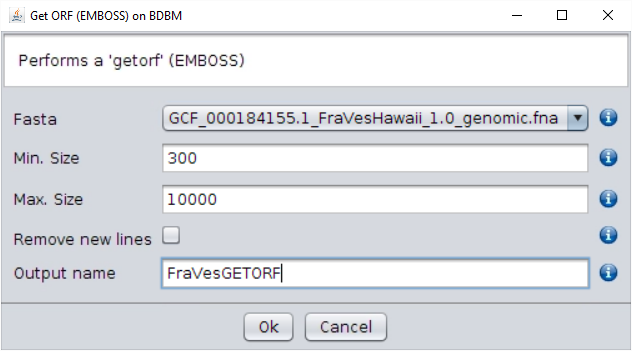
/fasta/nucleotidesfolder of the repository). - Select the Operations → Get ORF (EMBOSS) operation and use the default
parameters (we can use this option since, in plants, SFBB genes are always
intronless).
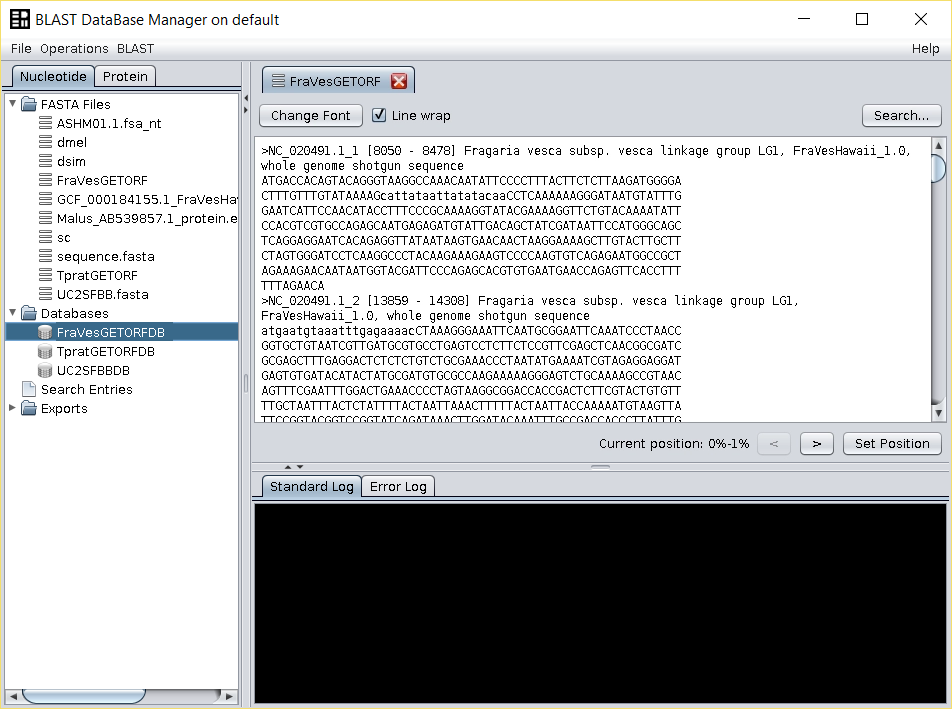
Give the name of your choice to the output (such asFraVesGETORF, the name used in this example). The output ORFs file will appear in theFASTA filesfolder of the repository tree.
The new file will contain all the ORFs that start with a methionine and that end with a codon or that have no stop codons untils the end of the sequence. - Build a BLAST-formatted database by right clicking on top of the
FraVesGETORFfile and by choosing the Make Blast Database operation. Choose a name for this nucleotide database, such asFraVesGETORFDB. - Download the Malus (AB539857.1) protein sequence from here (mirror).
- Remove the first 50 amino acids from the Malus (AB539857.1) protein sequence using your favourite plain text editor.
- Import the edited protein sequence FASTA file to BDBM using the File →
Import FASTA operation (or copy the file to the
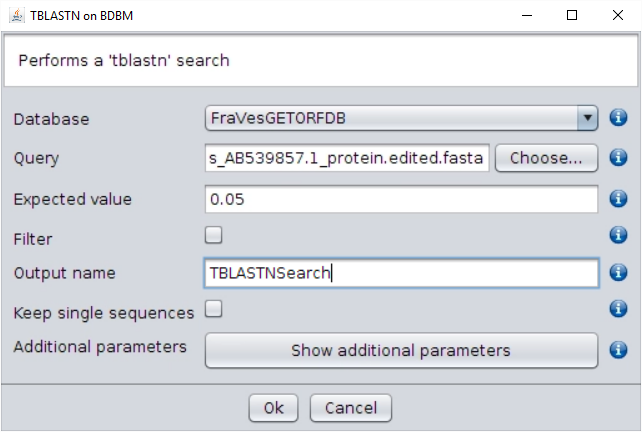
/fasta/proteinsfolder of the repository). - Perform a
tblastnsearch using the BLAST → TBLASTN (with external query) operation. Use as query the edited SFBB FASTA file, and give a name to the output. Two files are generated: the BLAST output file and a file with the sequences that show a hit in the BLAST output file (the SFBB-like sequences). Both are found in the folderExports.


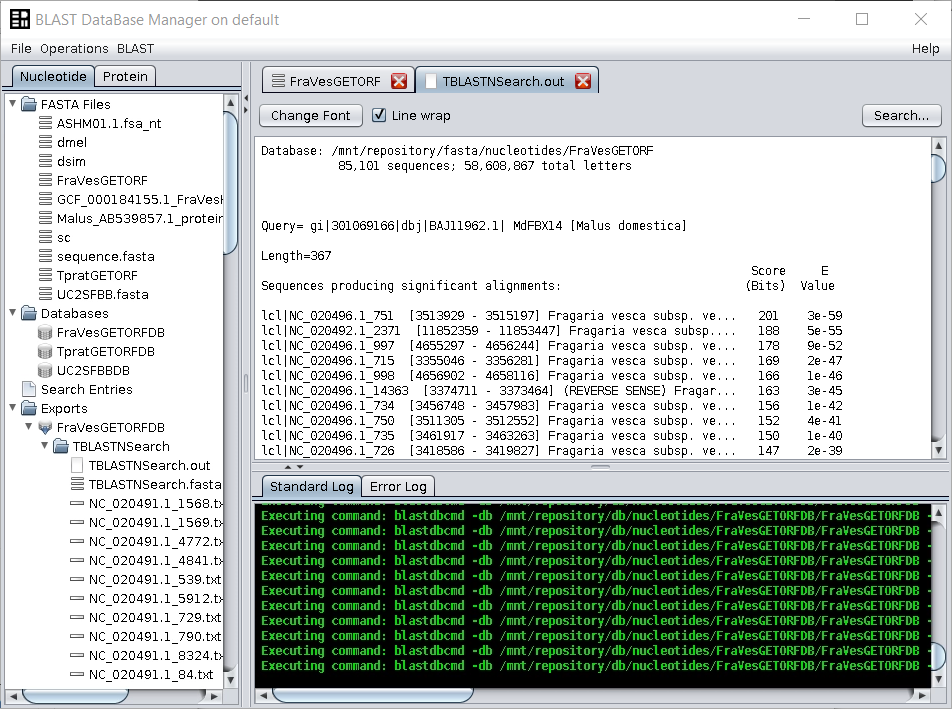
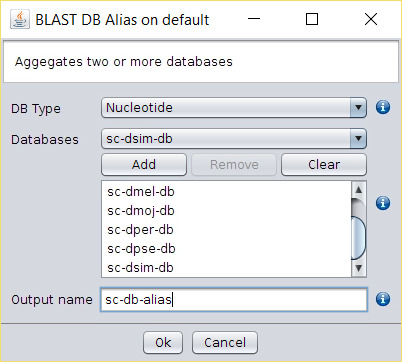
Final note
It should be noted that if the goal is to produce a FASTA file with SFBB nucleotide sequences from several species, then the Operations → Get ORF operation should be performed separately for each species, and the appropriate prefix added (using the Operations → Reformat FASTA operation) to each file so that we can track the species of origin for each sequence. BLAST databases should be created separately for each file, and an alias created (using the Operations → BLAST DB Alias operation), so that all databases of interest will be treated as a single one. tblastn searches can then be performed using the SFBB protein sequence of interest. This will produce a FASTA file with the SFBB nucleotide sequences from all species considered. Please note that when using the Operations → Get ORF operation erroneous CDS annotations can be obtained due to the presence of sequencing gaps, or errors in the original genome/transcriptome sequences, so doing some checks on the sequences (after aligning them, for instance) is advisable.
Use Case 3. Annotating alternative splicing isoforms
For many species, such as D. melanogaster, more than one coding sequence is described for each gene due to the use of alternative translation starts, and/or alternative splicing. For instance, for the D. melanogaster Sod gene, two coding sequences are available in FlyBase (462 and 504 bp long; link). Nevertheless, in the the closely related species D. simulans, only the 462 bp long coding sequence is described in FlyBase (link). BDBM can be used to quickly obtain the other form.
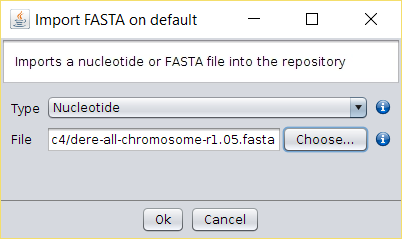
- First, we need to download the D. simulans genome assembly from here (mirror) and extract the contents.
- Then, download the D. melanogaster 504 bp Sod isoform from here (mirror).
- Import both FASTA files into BDBM using the File → Import FASTA
option (or copy the file to the
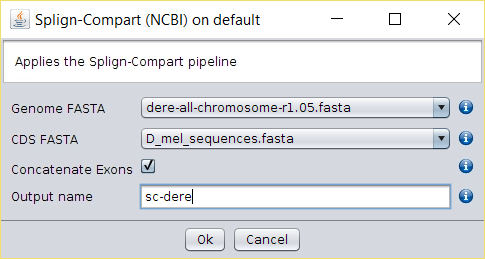
/fasta/nucleotidesfolder of the repository folder). - Running the Operations → Splign-Compart operation will produce a FASTA file with the D. simulans 504 bp isoform.
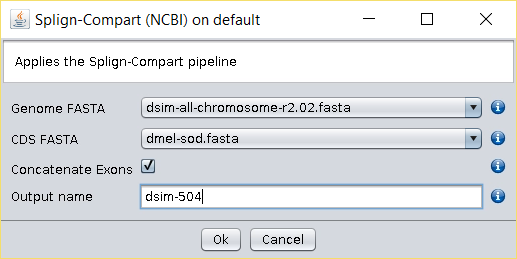
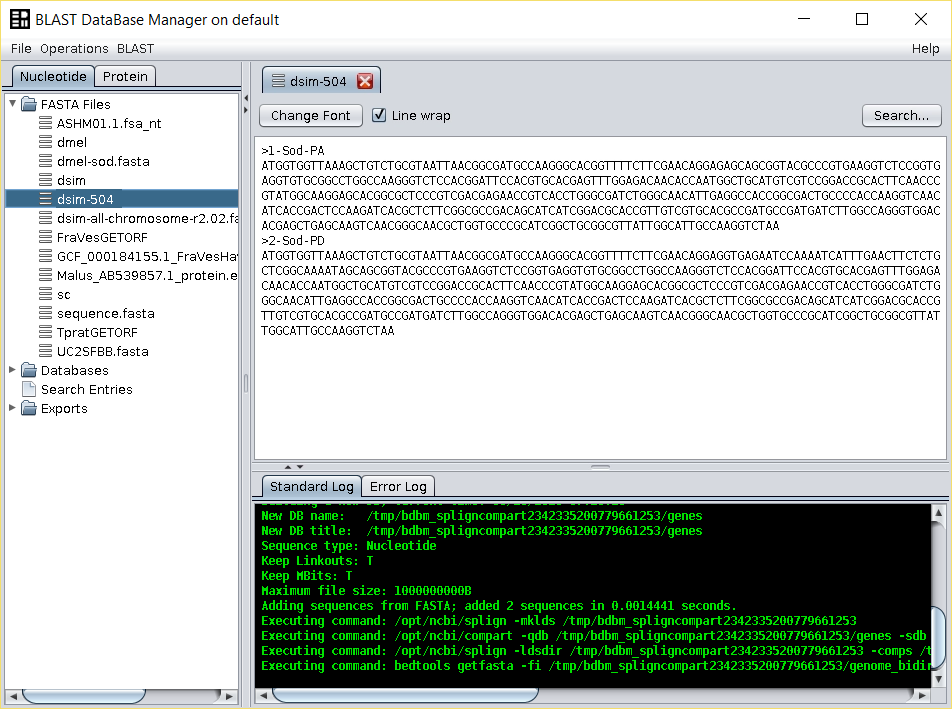
Final note
It should be noted that this operation can be done using multiple coding sequences at the same time, and that the header of the sequence used to produce a given annotation is transferred to the resulting FASTA file. Therefore, it is advisable to use the BDBM Operations → Reformat FASTA operation to produce a file that looks exactly as the researcher wants.
The Operations → Splign-Compart operation is not intended to be a gene annotation software, but rather a tool that allows the transference of a gene annotation available for a given species to another closely related species.
Use Case 4. Deriving alternative gene annotations
The availability of a genome annotation does not mean that CDSs are correctly annotated. When a gene annotation is very different in two closely related species, the Operations → Splign-Compart operation can be used to try to find an alternative gene annotation that would make the two gene annotations more similar.
In order to determine the feasibility of this approach, we can attempt to obtain a gene annotation for all 42 genes under the GO category germ cell development (i.e. alien, aret, bam, bgcn, Cul2, Cul5, CSN1b, CSN3, CSN4, CSN5, CSN6, CSN7, dpp, ecd, EcR, egh, fmr1, fs(1)Yb, gcl, gus, hyd, Jafrac1, Kay, lok, meiP26, nclb, Nop60B, nos, orb, osk, otu, out, pgc, pum, shg, SmB, Sxl, Tre1, tud, usp, wun2, and zpg) from 15 Drosophila genomes/transcriptomes (i.e. D. simulans, D. sechellia, D. yakuba, D. erecta, D. ficusphila, D. eugracilis, D. biarmipes, D. takahashii, D. suzukii, D. serrata, D. elegans, D. rhopaloa, D. kikkawai, D. ananassae, and D. bipectinata).
All 42 D. melanogaster protein coding genes under the GO category germ-cell development were retrieved from Flybase. For your convenience, you can download them all from here.
All but four (Jafrac1, nclb, pgc and usp) of these genes show introns in the coding region of the transcript studied. Therefore, in order to retrieve the corresponding CDS isoforms from the 15 non-melanogaster genomes and 1 transcriptome here considered, FASTA formatted genomes were downloaded from Flybase or from NCBI. You can find a list of the original URLs here, but, for your convenience, we have created a bundle with all the FASTA files, that you can download from here.- Download the 42 D. melanogaster protein coding genes described before.
- For your convenience, you can download the CDS isoforms from the 15 non-melanogaster genomes and 1 transcriptome bundle instead of using the original URLs.
- Extract the FASTA files from the compressed files.
- Import all the FASTA files to BDBM using the File → Import FASTA operation
(or copy the files to the
/fasta/nucleotidesfolder of the repository). - Run the Operations → Splign-Compart using one of the 15 non-melanogaster genomes and 1 transcriptome and the multi FASTA file with a single D. melanogaster CDS isoform for each of these 42 genes.
- Repeat the previous step for each of the 15 non-melanogaster genomes and 1 transcriptome.
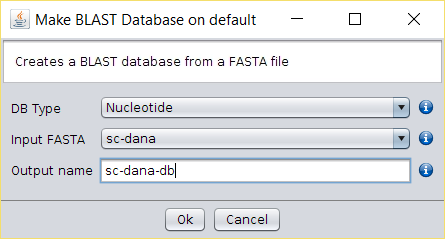
- Create a BLAST database for each of the 16 resulting FASTA files using Operations → Make BLAST Database.
- Run Operations → BLAST DB Alias to create and alias and treat all those databases as a single one.
-
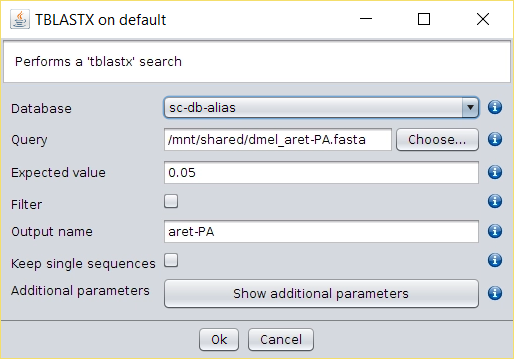
To be able to make a TBLASTX search with each of the 42 D. melanogaster protein coding genes we need to extract each one from the FASTA file
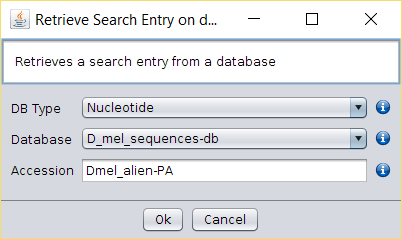
D_mel_sequences.fasta. You can achieve this by using the Operations → Retrieve Search Entry operation or your favourite text editor.Alternative 1. Using the Operations → Retrieve Search Entry operation-
Create a BLAST database for the
D_mel_sequences.fastafile using Operations → Make BLAST Database. -
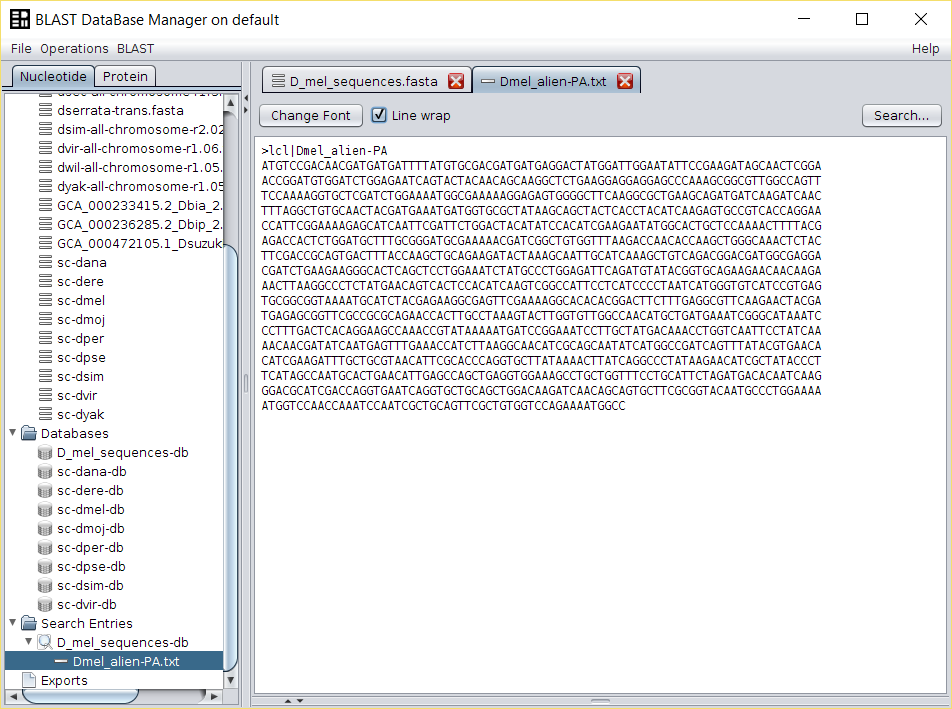
Open
D_mel_sequences.fastafile doing double click in the name of the FASTA file in the repository, select the name of one of the sequences and copy it to the clipboard. -
Run Operations → Retrieve Search Entry operation over the database
corresponding to the
D_mel_sequences.fastaFASTA file and use sequence name copied in the previous step. -
A new
Search Entrywill be created in BDBM with the contents of the selected sequence. - Repeat the previous steps for each of the 42 D. melanogaster protein coding genes.
- Perform a BLAST → TBLASTX search over the aliased database created in the previous steps using as query each search query created.
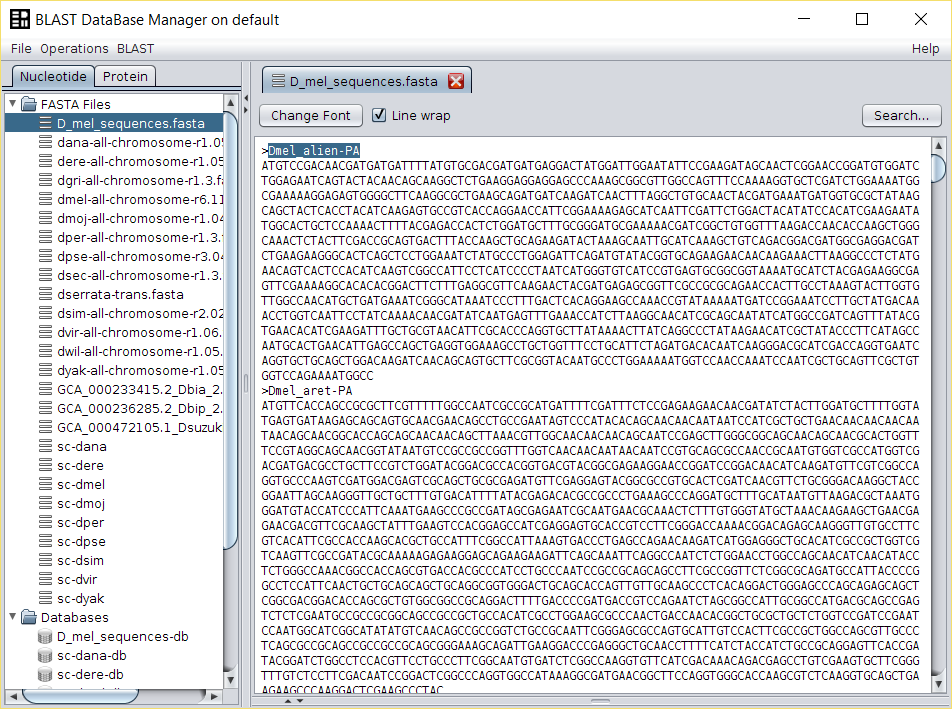
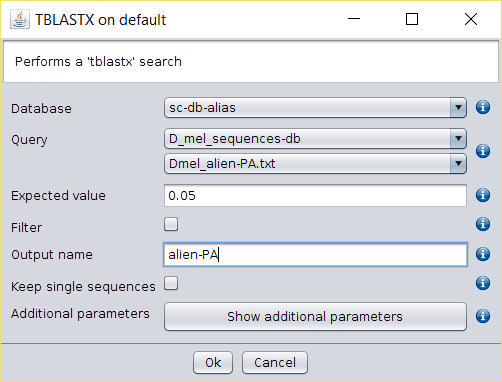 Alternative 2. Using your favourite text editor
Alternative 2. Using your favourite text editor-
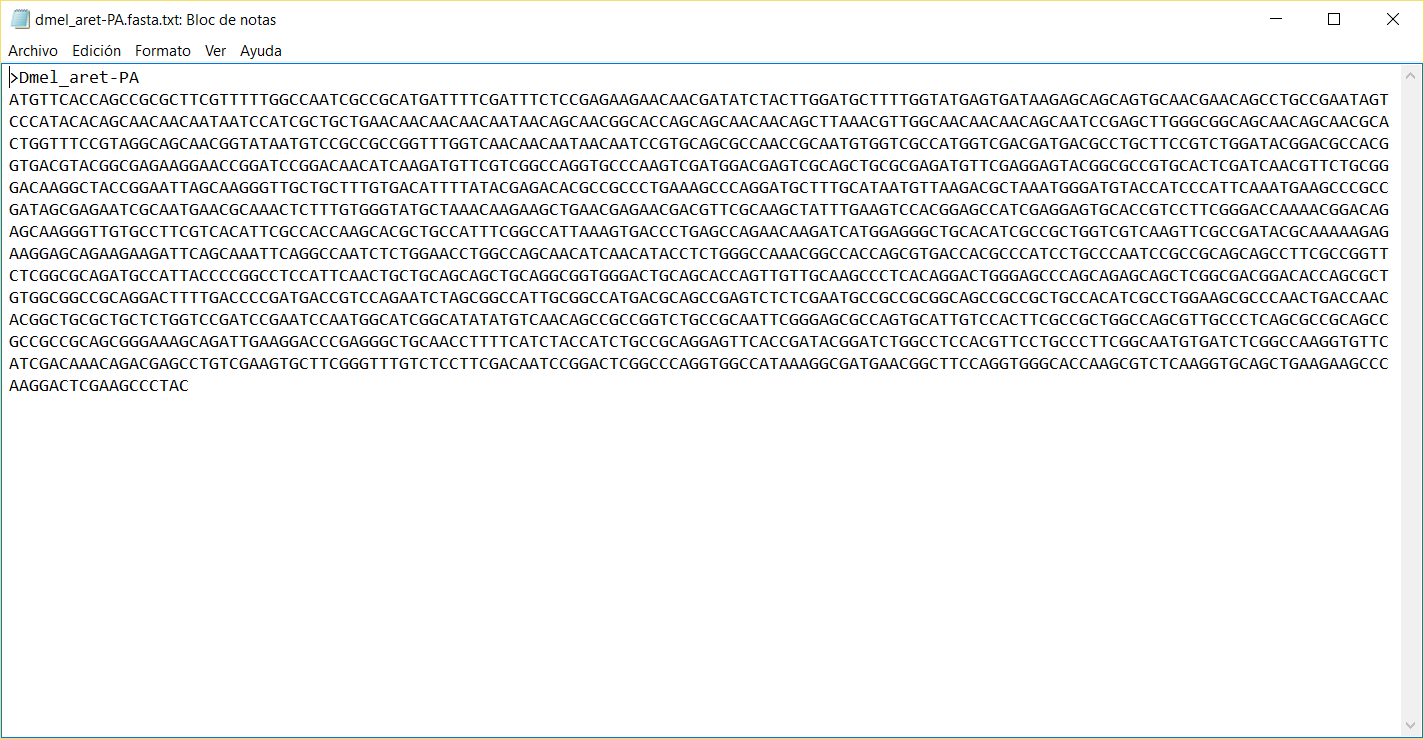
Open the
D_mel_sequences.fastaFASTA file using your favourite editor. - Copy each of the 42 D. melanogaster protein coding genes and paste them into separate FASTA files.
- Perform a BLAST → TBLASTX with external query search over de DB Alias created in the previous steps usign as query each of the files created in the previous step.
-
Create a BLAST database for the
- Check the resulting FASTA files to see if the CDS sequence is incomplete, or if there are obvious erroneous CDS annotations due to sequencing errors.
Note
As noted above, when a CDS annotation is performed, the original name of the sequence is replaced by the name of the sequence that is used to obtain a given CDS. This means that it is possible to use multiple CDSs at the same time as references, but on the other hand it means that in order to keep track of the species of origin, each genome/transcriptome must be used separately and the resulting CDSs prefixed with the species name using the prefix renaming mode from the Operations → Reformat FASTA operation.
Final note
One advantage of the Operations → Splign-Compart operation is that it is insensitive to frameshifts caused by sequencing errors because the annotation is based on similarity at the nucleotide level and presence of putative splice sites only. Therefore, the resulting annotation should always be checked to see if the resulting CDS sequence lengths are multiple of three. It should also be noted that fast evolving genes are unlikely to be annotated this way.

As stated above, the Operations → Splign-Compart operation is not intended to be a genome annotation software but rather a useful tool that allows the easy retrieval of high quality CDSs to be used in detailed analyses. Using this approach we could get a complete CDS annotation similar to that of D. melanogaster for 303 CDSs.
For those cases where we could not get an annotation when using the D. melanogaster CDS, we tried to obtain an annotation based on our annotations for non-melanogaster species or when using trusted CDS sequences from FlyBase for non-melanogaster species. The former approach resulted in a modest increase in the number of CDS sequences (26 CDSs, 8% increase) that could be used, but the use of the FlyBase D. ananassae CDSs allowed the retrieval of D. bipectinata CDSs for 40% (17/42) of the genes here considered (see Annotations).
When the CDSs that are produced using the Operations → Splign-Compart operation are compared with those available at FlyBase, for 88 cases the CDS obtained with BDBM is identical to that available at FlyBase, for 58 cases the CDS obtained with the Operations → Splign-Compart operation is more similar to the D. melanogaster CDS than the one available at FlyBase (mainly due to errors in homopolymer runs that cause frameshifts, or to the use of a different translation start), and for two cases the CDS obtained with the Operations → Splign-Compart operation is less similar to the D. melanogaster CDS than the one available at FlyBase.
Therefore, overall, the Operations → Splign-Compart operation implemented in BDBM indeed produces CDS annotations similar to those available in FlyBase, and thus can be used to produce alternative CDS annotations when checking for suspicious gene annotations. For 40 non-melanogaster CDSs, we used the sequence from FlyBase, since the provided annotation is similar to the annotated D. melanogaster CDS. These sequences will be used in Use Case 6. The files with all gene annotations are available at here.
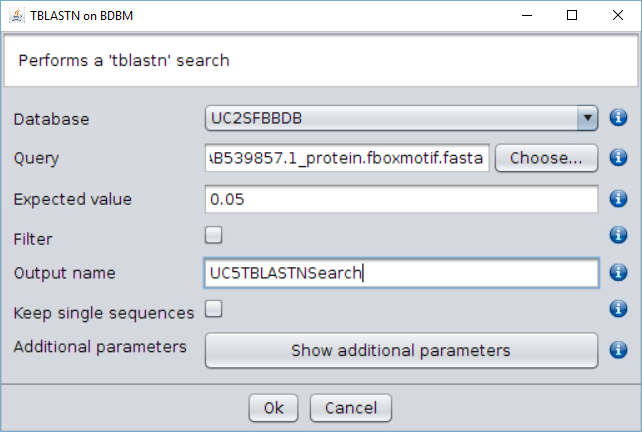
Use Case 5. Local BLAST
BDBM offers an easy to use local BLAST graphical interface. While it is true that researchers
must download the FASTA files of interest from existing databases and this is an extra
inconvenient step relative to the existing online BLAST databases, it is also true that, in
general, local BLAST searches are faster and that a much higher level of control regarding the
parameters used in the searches can be achieved. Particularly, BDBM BLAST operations include an
Additional parameters field that allows invoking all the arguments available for
BLAST, making it similar to running blast on the computer console, but with the
extra of automatically retrieving the sequences that show a hit in the blast search.
For instance:
- We could use the SFBB F-box gene annotation FASTA file produced in the Use Case 2 (Follow the instructions in Use Case 2 or download it from here).
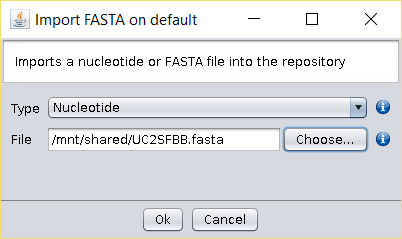
- Import the FASTA file to BDBM using the File → Import FASTA operation
(or copy the file to the
/fasta/nucleotidesfolder of the repository). -
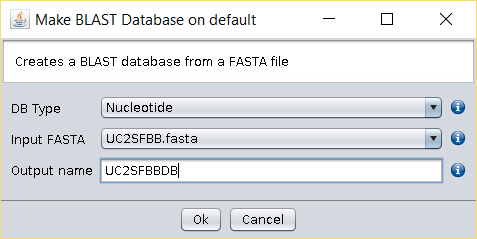
Build a BLAST-formatted database by right clicking on top of the
UC2SFBB.fastafile and by choosing the option Make Blast Database.
Choose a name for this nucleotide database, such asUC2SFBBDB. - Download the Malus (AB539857.1) protein sequence used in the Use Case 2 the from here (mirror).
- This time, remove all but the first 50 amino acids (the F-box motif) from the Malus (AB539857.1) protein sequence using your favourite plain text editor.
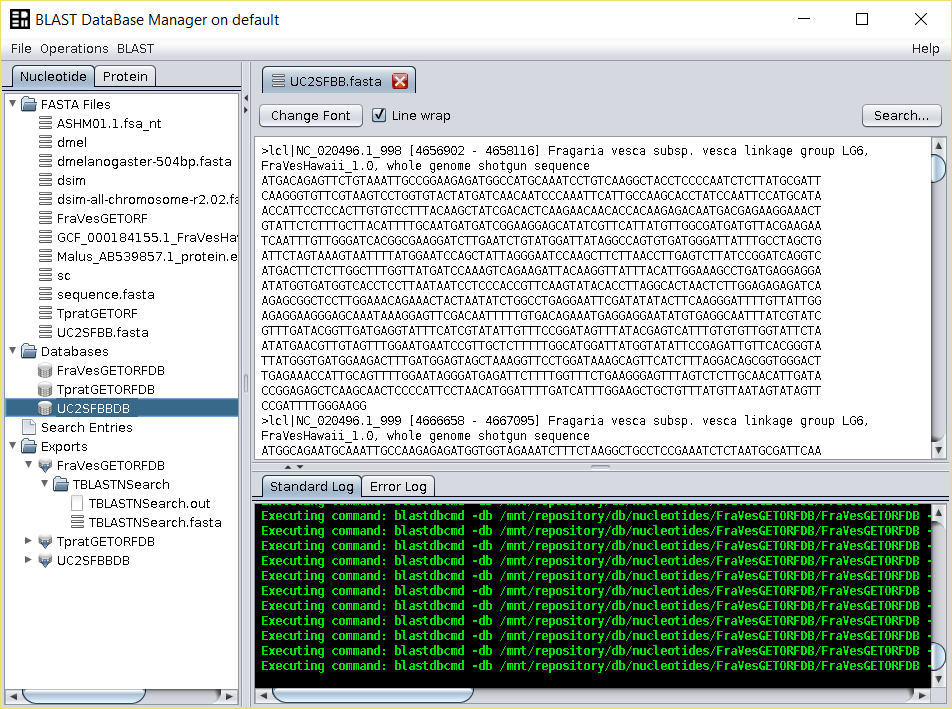
- Perform a
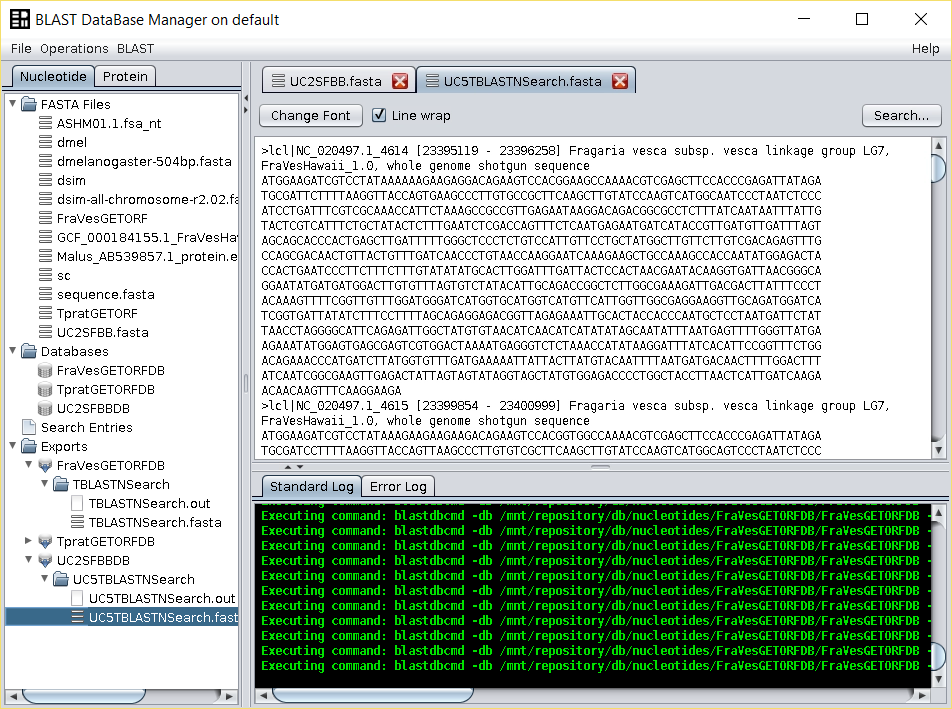
tblastnsearch over the nucleotide database (UC2SFBBDB) using the BLAST → TBLASTN (with external query) operation. Use as query the edited SFBB FASTA file in the previous step, and give a name to the output.
Two files are generated: the blast output file and a file with those entries that have a hit with the F-box motif, that are those that have at least a partial F-box region. Both are found in the folderExportsof the repository tree.

Final note
It is important to be careful when using custom additional parameters, as they can alter the format of the results, making them incompatible with other operations.
Use Case 6. Concatenating sequences for phylogenetic analyses
When performing phylogenetic analyses, researchers often concatenate sequences from different genes to increase the power to make inferences. Therefore, BDBM offers the possibility to concatenate sequences in FASTA format, as long as the sequence headers of the sequences to be concatenated are identical.
As an example of how easy and quick you can achieve this, we are going to merge 27 gene sequences of 6 different species of the Drosophila genus: D. melanogaster, D. sechellia, D. simulans, D. erecta, D. yakuba, and D. ananassae.
- Download the gene sequences from here.
- Extract the gene sequence files from the compressed file.
- Copy the extracted files to the
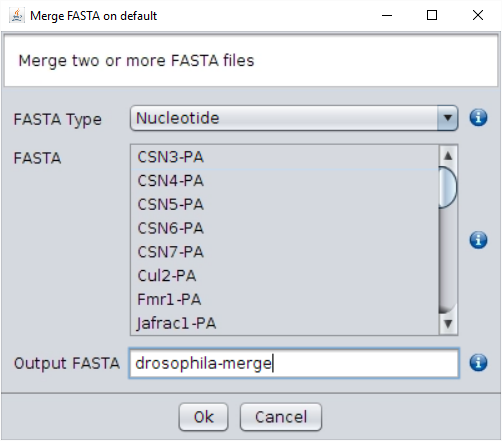
/fasta/nucleotidesfolder of the repository or import them using the File → Import FASTA operation. - Launch the Operations → Merge FASTA and select all the sequences.
Remember that you can select all the sequences in the list with the
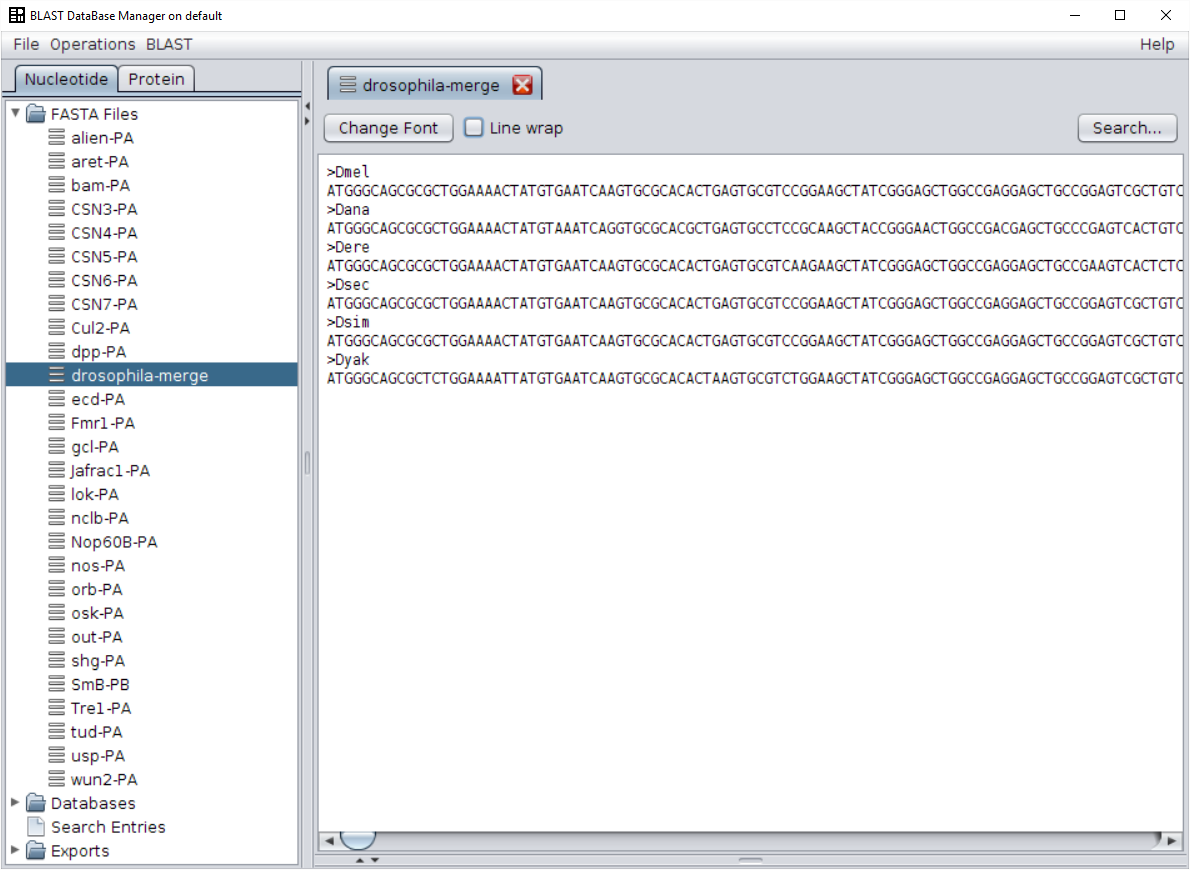
Ctrl + AorCmd + Acombination and that you can select or unselect single sequences pressingCtrlorCmdwhile clicking with the mouse. - Once you have selected all the sequences you want to merge, launch the execution by pressing the Ok button. The resulting FASTA file will contain six sequences, one for each species.
Final note
Together with the options implemented in BDBM that allow the quick annotation of genes (see above), this means that it is possible to quickly produce multigene FASTA files for phylogenetic analyses.
For instance, we could use some of the files of Use Case 4 (those for which a gene annotation is obtained for all the species of interest) to prepare a file to be used in phylogenetic analyses of the Drosophila genus using a large variety of computer programs.
Use Case 7. Transferring gene annotations between distantly related species
The ProSplign/ProCompart (NCBI) option is an alternative to Splign-Compart (NCBI). When using this option, protein reference sequences rather than CDSs (nucleotide) reference sequences are used. Since protein sequences change at a slower pace than nucleotide sequences, in principle the reference and target sequences can be more distantly related than when using the Splign-Compart (NCBI) option, but it is difficult to quantify how distantly related they can be. It should be noted that, when dealing with gene families, or genes that encode proteins with common protein motifs, partial annotations are usually obtained for many genes, although researchers are usually interested in full annotations only. The resulting CDS annotation is based on the homology to a given protein reference sequence, and thus may produce sequence annotations with lengths that are not multiple of three, if for instance, sequencing errors causing frameshifts are present in the genome to be annotated. Nevertheless, the existence of intron splicing signals at the exons 5’ and 3’ ends is taken into account. There will be no stop codon in the CDS annotation since the reference sequence is a protein. Splign-Compart (NCBI) runs considerably faster than ProSplign-Compart (NCBI), and thus users are advised to always try the Splign-Compart (NCBI) option first.
Here we attempt to obtain the Nicotiana attenuata PPCK1a CDS, using the Solanum tuberosum PPCK1a protein sequence (AF531415) as the reference.
- Download the Solanum tuberosum PPCK1a protein sequence (AF531415) from here.
- Import the AF531415.fasta file to BDBM using the File →
Import FASTA operation (choose type Protein) or copy the file to the
/fasta/proteinsfolder of the repository. - Download the Nicotiana attenuata genome assembly (GCA_001879085.1) from here and uncompress it in your computer.
- Import the GCF_001879085.1_NIATTr2_genomic.fna file to BDBM using the File →
Import FASTA operation (choose type Nucleotide) or copy the file to the
/fasta/nucleotidesfolder of the repository. - Launch the Operations → ProSplign/ProCompart (NCBI) operation and choose the GCF_001879085.1_NIATTr2_genomic.fna file as Nucleotides FASTA (the genome file), the AF531415.fasta as Query protein FASTA (the file with the Solanum tuberosum PPCK1a protein sequence) and a file output name
A large file with many partial annotations will be produced but only three full-length CDS sequences (822, 831 and 834 bp long) will be obtained, that can be identified as those long sequences that start with an ATG (start codon). If only the start or end of the CDS is missing users may try to obtain the full sequence by using the BDBM “Refine annotation” option, which is not needed for this example. The main results file is saved in the /fasta/nucleotides folder that is located in the specified repository folder. The first number on the header is an index that is followed by the name of the protein sequence used to obtain the annotation. The remaining information gives the possibility to link this file to two other files that will be saved in the /Export Files/nucleotides folder that is located in the specified repository folder (see below), and information on the name of the sequence that was annotated (see text after Header:). In the /Export Files/nucleotides folder, the file with the txt extension shows the output of the tblastx search used for the subsequent annotation, while the file with the fasta extension gives the genome region where the gene has been annotated, including the name of the target nucleotide sequence (see text after Header:). The correspondence between the two files is made by looking at the first four numbers in the file with the Fasta extension that must match the first, second, fourth and fifth number, respectively, in the file with the txt extension. It should be noted that a single reference sequence can give rise to more than one annotation if ProSplign cannot completely confidently align the reference and target sequences (those positions that are confidently aligned are labelled with an asterisk in the file with the txt extension).
